

Search
- Page Path
-
- HOME
- Search
- Review Article
- Genetics and Metabolism
- Development of orphan drugs for rare diseases
- Han-Wook Yoo
- Clin Exp Pediatr. 2024;67(7):315-327. Published online June 28, 2023
-

· Orphan disease is a rare disease, primarily affecting newborn and children. Vast majority of orphan diseases has genetic background.
· Orphan disease is individually rare. But as a whole, it is not rare, becoming a great socioeconomic burden.
· The diagnosis of rare genetic disease has been problematic, but recent progress of genome analysis technologies makes it faster and more precise.
· There are many unmet needs as to the curative treatment. However, the number of treatable rare diseases is growingly increasing owing to the development of biotechnology.
· Most orphan drugs are extremely expensive because of numer ous hurdles during the process of drug development as well as small number of patients.
- Original Article
- Endocrinology
- Comparison of effectiveness of growth hormone therapy according to disease-causing genes in children with Noonan syndrome
- Kyo Jin Jo, Yoo Mi Kim, Ju Young Yoon, Yeoun Joo Lee, Young Mi Han, Han-Wook Yoo, Hyang-Sook Kim, Chong Kun Cheon
- Clin Exp Pediatr. 2019;62(7):274-280. Published online December 3, 2018
-
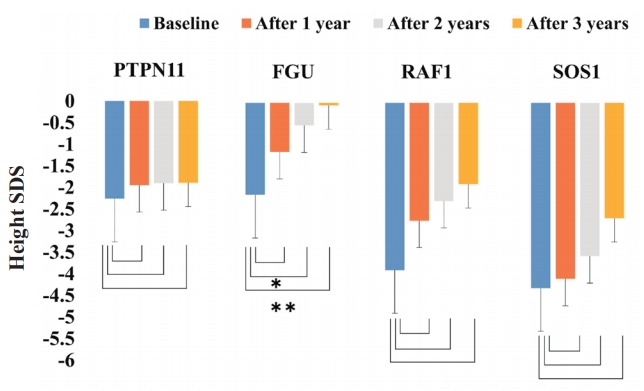
Purpose: To analyze the growth response to growth hormone (GH) therapy in prepubertal patients with Noonan syndrome (NS) harboring different genetic mutations. Methods: Twenty-three patients with prepubertal NS treated at Pusan National University Children’s Hospital between March 2009 and July 2017 were enrolled. According to the disease-causing genes identified, the patients with NS were divided into 4 groups. Three groups were...
- Review Article
- Endocrinology
- Management issues of congenital adrenal hyperplasia during the transition from pediatric to adult care
- Jin-Ho Choi, Han-Wook Yoo
- Clin Exp Pediatr. 2017;60(2):31-37. Published online February 27, 2017
-
Steroid 21-hydroxylase deficiency is the most prevalent form of congenital adrenal hyperplasia (CAH), accounting for approximately 95% of cases. With the advent of newborn screening and hormone replacement therapy, most children with CAH survive into adulthood. Adolescents and adults with CAH experience a number of complications, including short stature, obesity, infertility, tumor, osteoporosis, and reduced quality of life. Transition from...
- Case Report
- Genetics and Metabolism
- Long-term clinical course of a patient with mucopolysaccharidosis type IIIB
- Ja Hye Kim, Yang Hyun Chi, Gu-Hwan Kim, Han-Wook Yoo, Jun Hwa Lee
- Clin Exp Pediatr. 2016;59(Suppl 1):S37-S40. Published online November 30, 2016
-
Mucopolysaccharidosis type III (MPS III) is a rare genetic disorder caused by lysosomal storage of heparan sulfate. MPS IIIB results from a deficiency in the enzyme alpha-
N -acetyl-D-glucosaminidase (NAGLU). Affected patients begin showing behavioral changes, progressive profound mental retardation, and severe disability from the age of 2 to 6 years. We report a patient with MPS IIIB with a long-term follow-up...
- Endocrinology
- Phelan-McDermid syndrome presenting with developmental delays and facial dysmorphisms
- Yoon-Myung Kim, In-Hee Choi, Jun Suk Kim, Ja Hye Kim, Ja Hyang Cho, Beom Hee Lee, Gu-Hwan Kim, Jin-Ho Choi, Eul-Ju Seo, Han-Wook Yoo
- Clin Exp Pediatr. 2016;59(Suppl 1):S25-S28. Published online November 30, 2016
-
Phelan-McDermid syndrome is a rare genetic disorder caused by the terminal or interstitial deletion of the chromosome 22q13.3. Patients with this syndrome usually have global developmental delay, hypotonia, and speech delays. Several putative genes such as the
SHANK3 ,RAB ,RABL2B , andIB2 are responsible for the neurological features. This study describes the clinical features and outcomes of Korean patients with...
- Neurology
- Two cases of familial cerebral cavernous malformation caused by mutations in the
CCM1 gene - Im-Yong Yang, Mi-Sun Yum, Eun-Hee Kim, Hae-Won Choi, Han-Wook Yoo, Tae-Sung Ko
- Clin Exp Pediatr. 2016;59(6):280-284. Published online June 30, 2016
-
Cerebral cavernous malformation (CCM) is a vascular malformation characterized by abnormally enlarged capillary cavities without any intervening neural tissue. We report 2 cases of familial CCMs diagnosed with the
CCM1 mutation by using a genetic assay. A 5-year-old boy presented with headache, vomiting, and seizure-like movements. Brain magnetic resonance imaging (MRI) revealed multiple CCM lesions in the cerebral hemispheres. Subsequent...
- Original Article
- Genetics and Metabolism
- Identification of 1p36 deletion syndrome in patients with facial dysmorphism and developmental delay
- Go Hun Seo, Ja Hye Kim, Ja Hyang Cho, Gu-Hwan Kim, Eul-Ju Seo, Beom Hee Lee, Jin-Ho Choi, Han-Wook Yoo
- Clin Exp Pediatr. 2016;59(1):16-23. Published online January 22, 2016
-
Purpose The 1p36 deletion syndrome is a microdeletion syndrome characterized by developmental delays/intellectual disability, craniofacial dysmorphism, and other congenital anomalies. To date, many cases of this syndrome have been reported worldwide. However, cases with this syndrome have not been reported in Korean populations anywhere. This study was performed to report the clinical and molecular characteristics of five Korean patients with the...
- Case Report
- Successful sulfonylurea treatment in a patient with permanent neonatal diabetes mellitus with a novel
KCNJ11 mutation - Sung Yeon Ahn, Gu-Hwan Kim, Han-Wook Yoo
- Clin Exp Pediatr. 2015;58(8):309-312. Published online August 21, 2015
-
Permanent neonatal diabetes mellitus refers to diabetes that occurs before the age of 6 months and persists through life. It is a rare disorder affecting one in 0.2-0.5 million live births. Mutations in the gene
KCNJ11 , encoding the subunit Kir6.2, and ABCC8, encoding SUR1 of the ATP-sensitive potassium (KATP) channel, are the most common causes of permanent neonatal diabetes mellitus....
- Original Article
- Lowe syndrome: a single center's experience in Korea
- Hyun-Kyung Kim, Ja Hye Kim, Yoo-Mi Kim, Gu-Hwan Kim, Beom Hee Lee, Jin-Ho Choi, Han-Wook Yoo
- Clin Exp Pediatr. 2014;57(3):140-148. Published online March 31, 2014
-
Purpose Lowe syndrome is a rare, X-linked recessive disorder caused by mutations in the
OCRL gene. It involves multiple anatomic systems, particularly the eyes, central nervous system, and kidneys, and leads to profound growth failure and global developmental delay. This study evaluated the clinical and genetic characteristics of Korean patients with Lowe syndrome.Methods The clinical findings and results of genetic studies were...
- Case Report
- Chronic intermittent form of isovaleric aciduria in a 2-year-old boy
- Jin Min Cho, Beom Hee Lee, Gu-Hwan Kim, Yoo-Mi Kim, Jin-Ho Choi, Han-Wook Yoo
- Clin Exp Pediatr. 2013;56(8):351-354. Published online August 27, 2013
-
Isovaleric aciduria (IVA) is caused by an autosomal recessive deficiency of isovaleryl-CoA dehydrogenase (IVD). IVA presents either in the neonatal period as an acute episode of fulminant metabolic acidosis, which may lead to coma or death, or later as a "chronic intermittent form" that is associated with developmental delays, with or without recurrent acidotic episodes during periods of stress, such...
- Two cases of chronic pancreatitis associated with anomalous pancreaticobiliary ductal union and
SPINK1 mutation - Eun Sam Rho, Earl Kim, Hong Koh, Han-Wook Yoo, Beom Hee Lee, Gu-Hwan Kim
- Clin Exp Pediatr. 2013;56(5):227-230. Published online May 28, 2013
-
Chronic pancreatitis is a progressive inflammatory disease resulting from repeated episodes of acute pancreatitis that impair exocrine function and eventually produce endocrine insufficiency. Some causes of chronic pancreatitis appear to be associated with alterations in the serine-protease inhibitor, Kazal type 1 (
SPINK1 ), cationic trypsinogen (PRSS1 ), and cystic fibrosis-transmembrane conductance regulator (CFTR ) genes, or with structural disorders in the pancreaticobiliary ductal...
- X-linked recessive myotubular myopathy with
MTM1 mutations - Young-Mi Han, Kyoung-Ah Kwon, Yun-Jin Lee, Sang-Ook Nam, Kyung-Hee Park, Shin-Yun Byun, Gu-Hwan Kim, Han-Wook Yoo
- Clin Exp Pediatr. 2013;56(3):139-142. Published online March 18, 2013
-
X-linked recessive myotubular myopathy (XLMTM) is a severe congenital muscle disorder caused by mutations in the
MTM1 gene and characterized by severe hypotonia and generalized muscle weakness in affected males. It is generally a fatal disorder during the neonatal period and early infancy. The diagnosis is based on typical histopathological findings on muscle biopsy, combined with suggestive clinical features. We...
- Magnetic resonance imaging and spectroscopic analysis in 5 cases of Pelizaeus-Merzbacher disease: metabolic abnormalities as diagnostic tools
- Eun Lee, Mi-Sun Yum, Hae-Won Choi, Han-Wook Yoo, Su Jeong You, Eun-Hye Lee, Tae-Sung Ko
- Clin Exp Pediatr. 2012;55(10):397-402. Published online October 29, 2012
-
Pelizaeus-Merzbacher disease (PMD) is a rare, X-linked recessive disorder characterized by dysmyelination in the central nervous system. PMD results from deletion, mutation, or duplication of the proteolipid protein gene (
PLP1 ) located at Xq22, leading to the failure of axon myelination by oligodendrocytes in the central nervous system. PMD may be suspected when there are clinical manifestations such as nystagmus, developmental...
- Original Article
- Clinical and genetic characteristics of Gaucher disease according to phenotypic subgroups
- Ju-Young Lee, Beom Hee Lee, Gu-Hwan Kim, Chang-Woo Jung, Jin Lee, Jin-Ho Choi, Han-Wook Yoo
- Clin Exp Pediatr. 2012;55(2):48-53. Published online February 14, 2012
-
Purpose Gaucher disease is caused by a β-glucocerebrosidase (GBA) deficiency. The aim of this study is to investigate the clinical and genetic characteristics according to subtypes of Gaucher disease in the Korean population.
Methods Clinical findings at diagnosis,
GBA mutations, and clinical courses were reviewed in 20 patients diagnosed with Gaucher disease.Results Eleven patients were diagnosed with non-neuronopathic type, 2 with acute neuronopathic type,...
- Case Report
- Transient neonatal diabetes mellitus with macroglossia diagnosed by methylation specific PCR (MS-PCR)
- Hye Young Jin, Jin-Ho Choi, Gu-Hwan Kim, Han-Wook Yoo
- Clin Exp Pediatr. 2010;53(3):432-436. Published online March 15, 2010
-
Transient neonatal diabetes mellitus (TNDM) has been associated with paternal uniparental isodisomy of chromosome 6, paternally inherited duplication of 6q24, or a methylation defect at a CpG island of the ZAC or HYMAI gene. We experienced a case of TNDM in which the patient presented with hyperglycemia, macroglossia, and intrauterine growth retardation, caused by a paternally derived HYMAI. An 18-day-old... -
- Original Article
- Endocrine dysfunction after bone marrow transplantation during childhood and adolescence
- Hye Young Jin, Jin-Ho Choi, Ho-Joon Im, Jong-Jin Seo, Hyung-Nam Moon, Han-Wook Yoo
- Clin Exp Pediatr. 2010;53(3):420-427. Published online March 15, 2010
-
Purpose : Several complications can occur in patients who received bone marrow transplantation (BMT) during childhood and adolescence. This study aims to investigate endocrine dysfunctions after BMT so that better care can be provided to care for long-term survivors of BMT. Methods : One hundred patients (61 males, 39 females) were included in this study. Clinical parameters such as initial diagnosis,... -
- Case Report
- A case of Smith-Lemli-Opitz syndrome diagnosed by identification of mutations in the 7-dehydrocholesterol reductase (DHCR7) gene
- Mee Rim Park, Jung Min Ko, Chong-Keun Cheon, Gu-Hwan Kim, Han-Wook Yoo
- Clin Exp Pediatr. 2008;51(11):1236-1240. Published online November 15, 2008
-
Smith-Lemli-Opitz syndrome (SLOS) is a rare, autosomal recessive disease caused by an inborn error in cholesterol synthesis. Patients with this disease suffer from multiple malformations due to reduced activity of 7-dehydrocholesterol reductase (DHCR7), which increases 7-dehydrocholesterol (7DHC) and 8-dehydrocholesterol (8DHC) concentrations and decreases cholesterol concentration in body fluids and tissue. The SLOS phenotypic spectrum ranges from a mild disorder with... -
- Original Article
- Genotype and clinical features of Korean patients with methylmalonic aciduria and propionic aciduria
- Eun Hye Lee, Jung Min Ko, Jae-Min Kim, Han-Wook Yoo
- Clin Exp Pediatr. 2008;51(9):964-970. Published online September 15, 2008
-
Purpose : Methylmalonic aciduria (MMA) and propionic aciduria (PA) are inborn errors in the catabolism of branched-chain amino acids. The study was undertaken to investigate the genotypes and clinical features of Korean patients with MMA and PA. Methods : This study examined 12 patients with MMA and eight with PA. We analyzed various clinical features, laboratory findings, treatments, and neuro-developmental outcomes.... -
- Phenotype-genotype correlations and the efficacy of growth hormone treatment in Korean children with Prader-Willi syndrome
- Keun Wook Bae, Jung Min Ko, Han-Wook Yoo
- Clin Exp Pediatr. 2008;51(3):315-322. Published online March 15, 2008
-
Purpose : Prader-Willi syndrome (PWS) is a complex genetic disorder, caused by the deletion of the paternally derived 15q11-13 region or the maternal uniparental disomy of chromosome 15 (mUPD(15)). In this study, we compared phenotypic differences between those patients whose disease was caused by microdeletion and those caused by mUPD(15). In addition, a comparison of the efficacy of growth hormone... -
- Review Article
- Diagnosis of inherited metabolic disorders based on their diverse clinical features and laboratory tests
- Han-Wook Yoo
- Clin Exp Pediatr. 2006;49(11):1140-1151. Published online November 15, 2006
-
Inherited metabolic disorders are individually rare but as a whole, they are nor rare. Since Archibald Garrod introduced a concept of “inborn error of metabolism” or “chemical individuality”, more than 500 diseases are currently known, affecting approximately one in 500 newborns cumulatively. They frequently manifest with acute, life-threatening crisis that require immediate specific intervention or they present with insidious diverse... -
- Case Report
- A Case of Tay-Sachs Disease in Korea Diagnosed by Enzyme Assay and DNA Analysis
- Hyun-Seung Jin, Jin-Ho Choi, Han-Wook Yoo
- Clin Exp Pediatr. 2004;47(12):1360-1363. Published online December 15, 2004
-
Tay-Sachs disease is an autosomal recessive, neurodegenerative disorder that results from excessive storage of the cell membrane glycolipid, and GM2 ganglioside within the lysosomes of cells. This disease is caused by deficiency of the isoenzyme β-hexosaminidase A, produced in the endoplasmic reticulum. Patients with Tay-Sachs disease are characterized by normal motor development in the first few months of life, followed... -
- A Case of Lesch-Nyhan Syndrome
- Joon-Sung Kim, Jae-Seung Lee, Ha-Young Noh, Byung-Ju Kim, Young-Jong Woo, Jee-Min Park, Myung-Gwan Kim, Gu-Hwan Kim, Han-Wook Yoo
- Clin Exp Pediatr. 2003;46(5):505-509. Published online May 15, 2003
-
Lesch-Nyhan syndrome is an X-linked recessive disorder characterized by hyperuricemia, choreoathetosis, spasticity, mental retardation, and compulsive, self-injurious behavior. This disorder results from a complete deficiency of the purine salvage enzyme, hypoxanthine-guanine phosphoribosyl transferase(HPRT). We report here on a case of Lesch-Nyhan syndrome in a 1-year, 7-month-old male who presented with frequent vomiting, failure to thrive, and developmental delay. The diagnostic... -
- Fanconi-Bickel Syndrome Presented with Diabetes Mellitus and Galactosemia : Identification of a Novel Mutation in the GLUT2 Gene
- You-Jeong Kim, Sun-Hee Rim, Young-Lim Shin, Han-Wook Yoo
- Clin Exp Pediatr. 2001;44(10):1201-1205. Published online October 15, 2001
-
Fanconi-Bickel syndrome is a rare autosomal recessive disorder of the carbohydrate metabolism recently demonstrated to be caused by mutations in GLUT2, the gene for the glucose transporter protein 2 expressed in the liver, pancreatic β islet-cells, intestine and kidney. Typical clinical and laboratory findings of Fanconi-Bickel syndrome are hepatomegaly secondary to glycogen accumulation, glucose and galactose intolerance, fasting hypoglycemia, a... -
- Original Article
- Identification of Novel Mutations and Three Most Common Mutations in the Human ATP7B Gene of Korean Patients with Wilson Disease
- Han-Wook Yoo, Gu-Hwan Kim, Ji-Won Chung, Chang-Yeon Lee, Kyung-Mo Kim
- Clin Exp Pediatr. 2001;44(5):569-576. Published online May 15, 2001
-
Purpose : Wilson disease is an autosomal recessive disorder of copper transport, which is probably the most common inherited metabolic disorder in Korea. It is characterized by defective biliary excretion of copper and impairment in the corporation of copper into ceruloplasmin. In Wilson disease, synthesis of a defective copper transporting enzyme leads to the accumulation of copper in the liver, brain and kidney. The... -
- Case Report
- A Case of Korean Patient with Nonketotic Hyperglycinemia; Diagnosed Based on CSF Amino Acid Analysis and Magnetic Resonance Spectroscopy
- Kie-Young Park, Ai-Rhan Kim, Ki-Soo Kim, Soo-Young Pi, Tae-Sung Ko, Jung-Hee Lee, Han-Wook Yoo
- Clin Exp Pediatr. 2000;43(7):993-999. Published online July 15, 2000
-
Nonketotic hyperglycinemia is an extremely rare congenital metabolic disorder, which is caused by the lack of a glycine cleavage system. The onset of hyperglycinemic symptom is during the neonatal or early infant period. Progressing grave neuromotor dysfunction is one of the main symptoms. They include myoclonic seizure, hiccup, apnea, decreased deep tendon reflex, lethargy and coma. The prognosis is mostly very poor. Furthermore, there... -
- Original Article
- Characterization of Molecular Defects in Korean Families with Inherited Ornithine Transcarbamylase Deficiency and Their Genotype-Phenotype Correlations
- Han-Wook Yoo
- Clin Exp Pediatr. 1999;42(7):900-910. Published online July 15, 1999
-
Purpose : This study was undertaken to characterize molecular defects in Korean families with ornithine transcarbamylase(OTC) deficiency, correlate it with phenotype using in vitro expression study, and utilize it for making prenatal molecular diagnosis. Methods : To investigate molecular lesions resulting in OTC deficiency in 15 unrelated Korean families, the OTC genes of probands were amplified exon by exon and analyzed... -
- Case Report
- A Case of Ornithine Transcarbamylase Deficiency Successfully Treated with Protein Restriction and Living Related Liver Transplantation
- Bong Seong Kim, Kyung Mo Kim, Han-Wook Yoo, Sung Gyu Lee
- Clin Exp Pediatr. 1999;42(6):868-873. Published online June 15, 1999
-
Ornithine transcarbamylase deficiency(OTCD), the most common inborn error of the urea cycle, is inherited in X-linked manner. In affected hemizygote males, OTCD manifests hyperammonemic coma that often leads to death during the newborn period. Our patient was at high risk for inborn error of urea cycle metabolism, since his two elder brothers died a few days after birth due to... -
- Two Cases of Citrullinemia Presented with Strokes
- Hyun-Mi Kim, Jae-Bok Kim, Jung-Ho Kim, Sang-Jin Ba, Chong-Hyun Yoon, Han-Wook Yoo
- Clin Exp Pediatr. 1999;42(3):437-441. Published online March 15, 1999
-
Urea cycle disorders are characterized by encephalopathy, respiratory alkalosis, and hyperammonemia. A urea cycle disorder should be considered a diagnostic possibility in any patient regardless of age with occult encephalopathy. The most common central nervous system pathology of urea cycle disorder is cerebral edema. The cerebral edema is caused by astrocyte swelling secondary to hyperammonemia and intracellular glutamine accumulation. Strokes... -
- A Case of Type 1 Gaucher Disease Treated with Enzyme Replacement
- Jae-Bok Kim, Han-Wook Yoo
- Clin Exp Pediatr. 1998;41(11):1590-1595. Published online November 15, 1998
-
Type 1 Gaucher disease is one of the most common genetic lysosomal storage disease caused by the deficiency of glucocerobrosidase. Deficiency of this enzyme results in accumulation of glucoceramide in the macrophage and leads to hepatosplenomegaly, pancytopenia, bone damage and sometimes can be fatal. Recently, enzyme replacement has been considered as a major therapeutic strategy and about 2,000 patients have... -
- Original Article
- Molecular Genetic Diagnosis in Korean Patients with Myoclonic Epilepsy with Ragged Red Fiber(MERRF) Syndrome
- Tae-Sung Ko, Sang-Ahm Lee, Gheeyoung Choe, Han-Wook Yoo
- Clin Exp Pediatr. 1998;41(7):941-952. Published online July 15, 1998
-
Purpose : Myoclonic epilepsy with ragged red fiber(MERRF) syndrome is a disease of the mitochondrial encephalomyopathies, characterized by progressive myoclonus(action), epilepsy, cerebellar ataxia, intention tremor, muscle weakness, progressive dementia, sensorineural hearing loss and optic atrophy. Its inheritance is maternally inherited mitochondrial mutation, and its pathologic finding is characterized by ragged red fibers(RRF). Biochemically its defects are diverse. This study was... -
-

-
-

-

-
Impact Factor3.2
-
8.02023CiteScore94nd percentilePowered by




")