Identification of 1p36 deletion syndrome in patients with facial dysmorphism and developmental delay
Article information
Abstract
Purpose
The 1p36 deletion syndrome is a microdeletion syndrome characterized by developmental delays/intellectual disability, craniofacial dysmorphism, and other congenital anomalies. To date, many cases of this syndrome have been reported worldwide. However, cases with this syndrome have not been reported in Korean populations anywhere. This study was performed to report the clinical and molecular characteristics of five Korean patients with the 1p36 deletion syndrome.
Methods
The clinical characteristics of the 5 patients were reviewed. Karyotyping and multiplex ligation-dependent probe amplification (MLPA) analyses were performed for genetic diagnoses.
Results
All 5 patients had typical dysmorphic features including frontal bossing, flat right parietal bone, low-set ears, straight eyebrows, down-slanting palpebral fissure, hypotelorism, flat nasal roots, midface hypoplasia, pointed chins, small lips, and variable degrees of developmental delay. Each patient had multiple and variable anomalies such as a congenital heart defect including ventricular septal defect, atrial septal defect, and patent duct arteriosus, ventriculomegaly, cryptorchism, or hearing loss. Karyotyping revealed the 1p36 deletion in only 1 patient, although it was confirmed in all 5 patients by MLPA analyses.
Conclusion
All the patients had the typical features of 1p36 deletion. These hallmarks can be used to identify other patients with this condition in their early years in order to provide more appropriate care.
Introduction
1p36 deletion syndrome [MIM #607872] is one of the most common submicroscopic deletion syndromes, and its incidence is estimated as 1:5,000 to 1:10,00012). Patients with 1p36 deletion syndrome share common clinical features, such as variable degrees of developmental delay/intellectual disability and distinctive craniofacial appearances, including microbrachycephaly, straight eyebrows, deep-set eyes, flat nasal bridges, midface hypoplasia, hypo- or hypertelorism, laterally down-slanting palpebral fissures, pointed chins, large late-closing anterior fontanels, and low-set ears134). In addition, patients have multisystemic complications. Hypotonia and seizure are noted in approximately half the cases45). Seizure types are variable with abnormal encephalogram (EEG) recordings5). Brain imaging may detect structural abnormalities, including enlarged lateral ventricles, cortical atrophy, enlarged subarachnoid spaces, and white matter abnormalities, in up to 88% of patients345). These patients also experience cardiac problems, including cardiomyopathy and structural congenital heart defects in over 70% of cases: atrial septal defects (ASD), ventricular septal defects (VSD), patent ductus arteriosus (PDA), valvular anomalies, tetralogy of Fallot, and Ebstein anomaly34). Opthalmologic abnormalities are noted in half the patients: strabismus, refractive errors, nystagmus, unilateral cataracts, retinal albinism, and visual inattentiveness. Hearing impairment (47%), conductive or sensorineural, may be revealed on auditory examination3). Skeletal findings, including delayed bone age, rib anomalies, lower limb asymmetry, scoliosis, and congenital hip dysplasia, may also develop3). Genitourinary anomalies and feeding difficulties are also observed, and endocrinologic evaluations may reveal hypothyroidism34).
Although known as one of the most common microdeletion syndromes, cases of this syndrome have not been reported in the Korean population. In the current study, the clinical and molecular characteristics of 5 Korean patients with 1p36 deletion syndrome are described. Our experience may provide a better understanding of this syndrome to help general clinicians identify more cases and provide more appropriate care.
Materials and methods
1. Patients
A total of 5 patients (3 males and 2 females) were identified as having 1p36 deletion syndrome at Asan Medical Center Children's Hospital, Seoul, Korea, between March 2005 and July 2014. Their detailed clinical findings, comprising gender, age at diagnosis, facial dysmorphism, and the surveillances of multiorgan functions including the heart, endocrine system, and brain, were reviewed with growth and developmental patterns.
2. Genetic testing
Informed consent was obtained from the parents of each patient. Chromosome analysis was performed using each patient's leukocytes in metaphase by conventional G-banded techniques at the level of 550 bands. Multiplex ligation-dependent probe amplification (MLPA) analyses were done using two types of SALSA Reference Kit, P064 and P245 (MRC Holland, Amsterdam, the Netherlands) according to the manufacturer's instructions. Amplified products were separated using ABI3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) and analyzed by Gene Mapper Software (Applied Biosystems). The probemixes of P064-B3 and P245-A2 were used before 2013, and those of P064-C1 and P245-B1 have been used since 2013 according to the version updates. P064-B3 and P064-C1 contain 43 and 52 MLPA probes with amplification products, respectively, corresponding to the TNFRSF18, TNFRSF4, SCNN1D, GNB1, SKI, PANK4, GABRD, PEX10, and ACTRT2 genes located at the 1p36.33–1p32 regions. P245-A2 and P245-B1 contain 49 and 50 different MLPA probes with amplification products, respectively, corresponding to the TNFRSF4, GNB1, and GABRD genes located at the 1p36 region. The results included informative headers, electropherograms, ratio plots, validation boxes, and report tables. In the current study, we displayed the ratio plot of a specific gene. Green dots represent normal alleles, whereas red dots represent abnormal alleles that were deleted (Fig. 1).
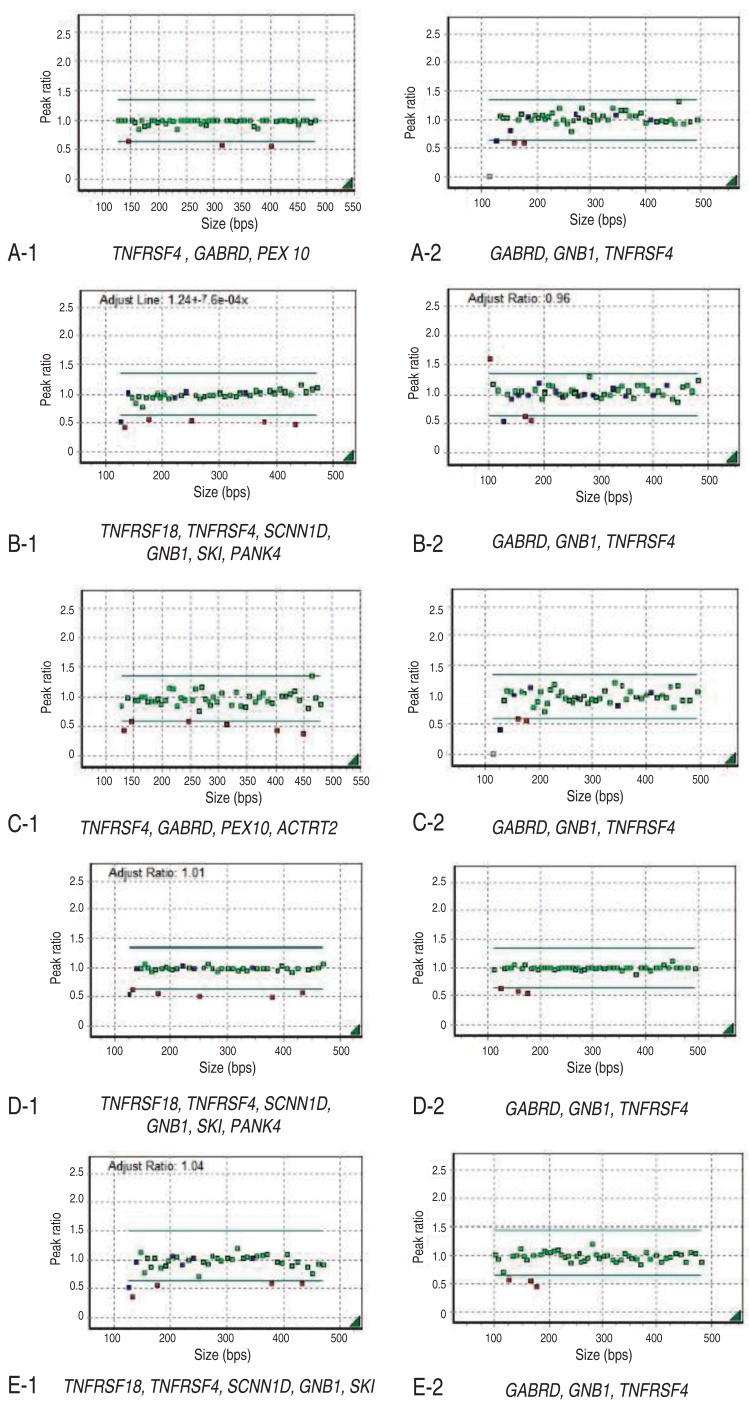
Results of multiplex ligation-dependent probe amplification analysis.
To exclude Prader-Willi syndrome (PWS) in case 1 and case 4, fluorescence in situ hybridization (FISH) analysis was done using probes for the SNRPN gene at 15q11-13. To exclude Fragile X syndrome (FXS) in case 4, triplet primed polymerase chain reaction (AmplideX, AsuraGen, Austin, TX, USA) was done to detect expanded trinucleotide (CGG) repeats (AmplideX), and capillary electrophoresis was done using ABI 3130 Genetic Analyzer (Applied Biosystems) to confirm abnormal CGG amplification of the FMR1 gene.
Results
The clinical and genetic findings of each patient are summarized in Table 1.

Clinical characteristics of patients with the 1p36 deletion syndrome
1. Case 1
The patient was the third baby of nonconsanguinous Korean parents. She was born after 41 gestational weeks with a birth weight of 2,750 g (standard deviation [SD], -1.8), a length of 50.4 cm (SD, -0.4), and a head circumference of 33.3 cm (SD, -1.4). Pregnancy, labor, and vaginal delivery were uneventful. At 3 months of age, generalized tonic-clonic seizures developed, although EEG and brain magnetic resonance imaging (MRI) were normal. VSD, ASD, and PDA were also found on echocardiography and were surgically corrected at 1 year. Dysmorphic facial features were noted, including hypotelorism, straight eyebrows, midface hypoplasia, and a pointed chin. She could stand alone only after age 2 years and began to walk alone at 3 years, but she could not say any meaningful words. The results of auditory and ophthalmologic evaluations were normal. At age 6 years, her body mass index (BMI) was over the 97th percentile for her age, indicating severe obesity. At age 8, she was diagnosed with type 2 diabetes mellitus (DM). Scoliosis was found at age 10 years.
Her karyotype was 46,XX. FISH analysis for SNRPN at 15q11-q13 to rule out PWS was normal. However, MLPA analysis revealed haploinsufficiency of TNFRSF4, GABRD, PEX 10, and GNB1 in the 1p36 region (Figs. 1A, 2).
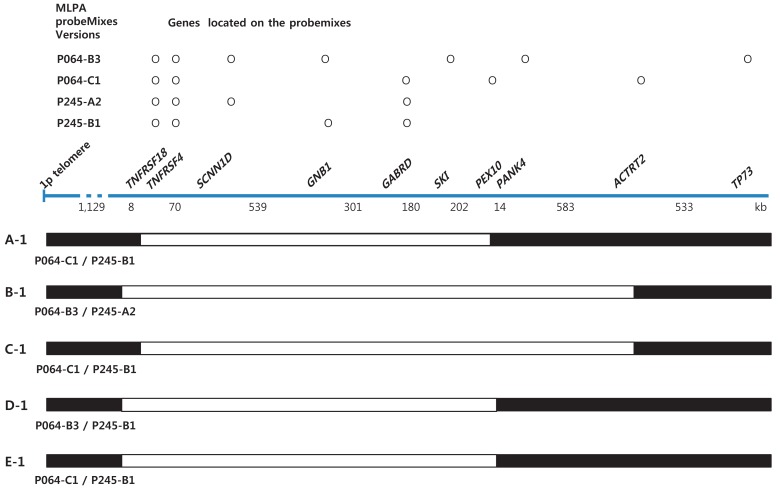
Schematic of the deleted genomic region in patients according to multiplex ligation-dependent probe amplification results.
Rehabilitation therapy has been required for her delayed development. Her DM has been well controlled with a metformin. The surgical correction was taken into consideration for progressive scoliosis.
2. Case 2
This patient was the first baby of nonconsanguineous Korean parents. He was born after 31 weeks of gestation by emergent Cesarean section due to maternal preeclampsia. His birth weight was 1,160 g (SD, -1.2), body length was 41.5 cm (SD, +0.4), and head circumference was 28.5 cm (SD, 0). His family members were healthy. Diffusely mild bilateral ventriculomegaly was detected at birth, which was non-progressive. Surgical closure was performed for VSD 60 days after birth. Dysmorphic features included frontal bossing, a flat nasal root, small lips, and low-set ears. He could control his head 5 months after birth and roll over at 10 months. At 1 year, he could sit with support. He could speak only a few words at 2 years. Global developmental delays, indicative of the level of 7–9 months of age, were noted on the Korean Infant and Child Development Test performed at age 1 year 4 months. Auditory, ophthalmologic, and thyroid function tests were normal.
His karyotype was 46,XY. 1p36 deletion was confirmed by MLPA analysis showing deletions at the TNFRSF18, TNFRSF4, SCNN1D, GNB1, SKI, PANK4, and GABRD genes (Figs. 1B, 2). Occupational and speech therapy have been given for his motor and speech delay.
3. Case 3
This patient was the first baby of nonconsanguinous Korean parents. She was born after 40 weeks of gestation weighing 2,580 g (SD, -1.9) and measuring 48 cm (SD, -1.1) with a head circumference of 34 cm (SD, -0.5). In the antenatal period, fetal ultrasonography (US) revealed ventriculomegaly, but chromosome analysis using amniotic fluid was 46,XX. Brain US at birth confirmed lateral ventriculomegaly, which was progressive in size on follow-up evaluation at 4 months of age. She received surgical ligation for PDA at age 2 months. She was hypotonic, and could control her head at 6 months and sit with support at 1 year. She had craniofacial dysmorphic features, including midface hypoplasia, frontal bossing, down-slanting palpebral fissures, and a flat right parietal bone. A thyroid function test and ophthalmologic evaluation were normal, but peripheral conduction defect was found in the left ear, for which a hearing aid was prescribed in the right ear. MLPA analysis revealed deletion of the TNFRSF4, GABRD, PEX 10, ACTRT2, and GNB1 genes in the chromosomal region 1p36 (Figs. 1C, 2).
She expired due to streptococcus pneumoniae sepsis at 2 years of age.
4. Case 4
The patient was the first baby of nonconsanguinous Korean parents. He was born after 40 weeks of gestation with a birth weight of 2,370 g (SD, -2.8). The prenatal evaluation was unremarkable and his perinatal period was uneventful. Developmental delays were also noted: he could walk alone only after 21 months of age and spoke two-word sentences after 2 years. His intelligence quotient score was 60 points, indicative of mild intellectual disability. He had a characteristic craniofacial appearance, including hypotelorism, a flat occiput, and a small mouth. Auditory and ophthalmologic examinations were normal. His thyroid function was normal, whereas hypercholesterolemia and hypertriglycemia were noted at age 3 years along with severe obesity (BMI>97th percentile). Cardiologic and neurologic examinations were normal.
His karyotype was 46,XY. A FISH study for PWS and genetic testing for FXS were normal, but MLPA analysis confirmed the deletion of the TNFRSF18, TNFRSF4, SCNN1D, GNB1, SKI, and GABRD genes at 1p36 (Figs. 1D, 2).
5. Case 5
This patient was the first baby of nonconsanguineous Korean parents. He was born after 39 weeks of gestation weighing 2,790 g (SD, -1.3) after an uncomplicated pregnancy. His family members were healthy. Cardiac VSD and ASD were detected and surgically corrected at 2 months of age. He had dysmorphic features, including a large anterior fontanel and midface hypoplasia. In addition, gastroesophageal reflux, bilateral cryptorchidism, and clinodactyly were found. Developmental delays were noted: he could only stand with support at 2 years of age and could not speak any meaningful words. Auditory examination was normal.
Chromosome analysis revealed 46,XY,del(1)(p36.3), and MLPA analysis revealed haploinsufficiency of the TNFRSF18, TNFRSF4, SCNN1D, GNB1, SKI, and GABRD genes at 1p36 (Figs. 1E, 2).
Discussion
1p36 deletion syndrome is a recently recognized disorder, with the first genetically confirmed case having only been published by Biegel at al.6) in 1993. In the current study, we described the general clinical characteristics and genetic findings of five Korean patients.
As in previously reported cases, our patients shared the common clinical features of patients with 1p36 deletion (Table 2)37). Most patients with 1p36 deletion are bone small for gestational age, as were all five patients in our study (mean, 2,306±638 g; normal range, 1,160–2,780 g). All patients with 1p36 deletion have global development delays and dysmorphic features134), likewise our patients had moderate-to-severe mental or cognitive impairment. In addition, the characteristic facial features of 1p36 deletion, including midface hypoplasia, hypotelorism, frontal bossing, small lips, a pointed chin, low-set ears, a large anterior fontanel, and microcephaly, were noted in our patients. Congenital heart defects have been reported in 71% of cases3), while 4 of our patients had VSD (60%), PDA (40%), and ASD (20%). Seizure and structural brain anomalies have been observed in a substantial number of 1p36 deletion syndrome patients34). In our cases, seizure was noted in 1 patient (20%) and lateral ventriculomegaly in 2 patients (40%). Ophthalmologic problems and hearing impairment have been reported in 52% and 47% of patients, respectively3), while only 1 patient in our study experienced sensorineural hearing loss. Other congenital anomalies, such as bilateral cryptorchidism (20%) and scoliosis (20%), were also found. In terms of endocrinologic findings, congenital hypothyroidism is common34). However, thyroid function in all the patients described here was normal.

Frequency of clinical findings in 5 patients compared to those commonly reported in 1p36 deletion
Although most patients with 1p36 deletion are born small for gestational age, obesity with hyperphagia and DM are also reported in a small number of patients as they grow up891011). In the current report, 2 patients (40%) had obesity and hyperchole sterolemia, and 1 patient was diagnosed with type 2 DM. In previous reports, a few regions were suggested as candidate regions for hyperphagia and obesity. In particular, PRKCZ, located between 2Mb and 3Mb from the 1p telomere, may be associated with obesity because this gene is involved in carbohydrate or lipid metabolism, or insulin signaling7). It was thus expected to be deleted in both obese patients in our study. It was also expected that the other three patients would have deletion at PRKCZ (Fig. 2) but they did not. Therefore, it is suggested that genetic or environmental factors more likely contribute to the development of obesity and DM. It is well known that children with intrauterine growth restriction grow more rapidly than normal babies but usually do not become overweight12). However, a subset of patients may become overweighigt and obese at adolescent and early adult periods13). Previous studies observed that obesity was found exclusively in female patients with 1p36 deletion who showed growth restriction during the fetal period14). However, in our study, both genders, one female and one male, were affected. Further study among a large cohort with 1p36 deletion is required to validate the high risk of obesity in 1p36 deletion patients (female patients, in particular).
Because patients with 1p36 deletion show hypotonia and hyperphagia with obesity and DM, which are also characteristic features of patients with PWS, some patients with 1p36 deletion may be misdiagnosed as having PWS. In our study, two patients tested negatively for PWS. Therefore, our experience indicates that 1p36 deletion should be considered in patients with overlapping features of PWS but with no deletion of the SNRPN gene at 15q11–13. FXS is one of most common genetic diseases with a range of developmental delays as well as typical facial features15). We performed molecular genetic testing of the FMR1 gene because this syndrome was suspected in case 4.
1p36 deletion syndrome comes from the truncation of the distal tip of the short arm of chromosome 1 due to terminal deletion, interstitial deletion, derivative chromosome, and complex rearrangement16). More than 60% of patients develop deletion de novo in the subtelomeric region of maternally inherited chromosome 116). While approximately 40% of all breakpoints occur 3.0–5.0 Mb from the 1p telomere, the majority of breakpoints are clustered 4.0–4.5 Mb from the 1p telomere16). However, the deletion sizes and breakpoints are variable, and no clear correlations between deletion type and size and the severity of clinical features have been revealed41617).
It has been suggested that several genes in this region are involved in the development of some phenotypes of 1p36 deletion syndrome. The haploinsufficiency of SKI (MIM164780) and PEX10 (MIM602859) genes may be associated with dysmorphic facial features, including cleft lip or palate, hypotonia, and developmental delays1819). The KCNAB2 (MIM601142) and GABRD (MIM137163) genes may also be involved in the neurological manifestations, including seizure2021). The defects of MMP23A (MIM 603320) and MMP23B (MIM 603321) have been suggested to cause large, late-closing anterior fontanels22).
Although the deletion of these genes has been implicated in the development of some anomalies, correlations between the haploinsufficiency of a gene and its corresponding phenotype have not been evident in general417). Then again, the altered expression of a few genes close to the deleted segments could also be involved in the phenotypic variability, as suggested by Redon et al.23), or a positional effect on one or more genes along the 1p36 region rather than classic contiguous gene deletion might have an additional effect on phenotype23).
For the diagnosis of 1p36 deletion syndrome, cytogenetic analyses, including conventional karyotyping, FISH, microarray comparative genomic hybridization (array CGH), and MLPA analysis, can be conducted. Conventional karyotyping only detects a large deletion over 5Mb and unbalanced translocation, and may fail to identify microdeletions, as noted in the majority of patients (4 out of 5) described here. The FISH test only detects the haploinsufficiency of 1 or 2 genes, whereas MLPA analysis covers multiple genes at 1p36 and is a more suitable test for detecting microdeletion. However, because the MLPA probemix used in the current study includes the genes located on the terminal portion of chromosome 1, MLPA cannot reveal interstitial deletion nor give the detailed information about deletion size, either. On the other hand, array CGH can analyze all the genomic regions of human chromosomes and can therefore be recommended as a confirmatory test to diagnose 1p36 deletion syndrome. However, this test is not commercially available in Korea at present, hence the MLPA test was applied in the current study.
In terms of genetic counseling, genetic testing is recommended for the parents of a patient with 1p36 deletion even though both parents may have normal phenotypes, because either parent can have the deletion without clinical manifestations due to the lack of penetrance of the deletion. For a child with an unbalanced translocation at 1p36, both parents should also be screened to determine whether either parent has a balanced translocation at 1p36 that was passed on to the offspring. If either parent is identified be a carrier of the chromosomal abnormality, close relatives of the carrier should also be screened for genomic abnormalities. Due to the variable degrees of phenotypic severity among patients, clinicians should be careful during genetic counselling about future family planning to the families with 1p36 deletion.
Management of this syndrome comprises a multidisciplinary approach. Along with growth and developmental assessments, systemic surveillance is required: echocardiogram, EEG, brain MRI, ophthalmologic examination, audiologic examination, thyroid function testing, palatal evaluation, and swallowing function studies. Appropriate general developmental assessments should include speech, physical, and behavioral functions. The mainstay of treatment is supportive management, as required, depending on each abnormality. Early rehabilitation therapy for global developmental delays is essential.
In conclusion, as the first report describing the clinical and molecular features of patients with 1p36 deletion syndrome in the Korean population, the current study indicates that the typical dysmorphic appearances and congenital multiple organ anomalies of 1p36 deletion were shared among our patients, but variable degrees of psychomotor developmental delay were noted. To identify more cases, molecular genetic testing such as array CGH is required. With early surveillance and the management of neurodevelopmental development in particular, more can be done to improve the clinical course of these patients.
Acknowledgment
This study was supported by a grant from the National Research Foundation of Korea, funded by the Ministry of Education, Science, and Technology (NRF-2011-0019674).
Notes
Conflicts of interest: No potential conflict of interest relevant to this article was reported.