


Introduction
Reports of constitutional ring chromosome 22, r(22) are rare. The majority of cases with r(22) are formed de novo, but there are a few reports of familial transmission1). A breakpoint resulting in the loss of the short arm and satellite material has few clinical consequences, whereas a breakpoint on the long arm, which can vary in size, can affect phenotypic expression depending on the size of the deletion2). Individuals with r(22) present with most features common to 22q13 deletion syndrome2,3). The phenotypes of individuals with r(22) can further be affected by the continuously evolving mosaicism, caused by the mitotic instability of the ring chromosome. Sister chromatid exchanges during mitosis can lead to the formation of dicentric or interlocked rings and subsequent aneuploidy or rearrangements within the chromosome4,5). However, mitotic instability in r(22) is a rare occurrence2,3). Central nervous system (CNS) atypical teratoid/rhabdoid tumors (ATRT) are rare, highly malignant tumors primarily occurring in young children under 3 years of age. The majority of ATRT cases display genetic alterations of SMARCB1 (INI1/hSNF5), a tumor suppressor gene located on 22q11.2, resulting in loss of INI1 protein. INI1 protein is ubiquitously expressed in the nuclei of all normal cells and can be identified using immunohistochemistry. Subsequent loss of INI1 protein expression comprises a relatively specific and sensitive diagnostic marker for ATRT6).
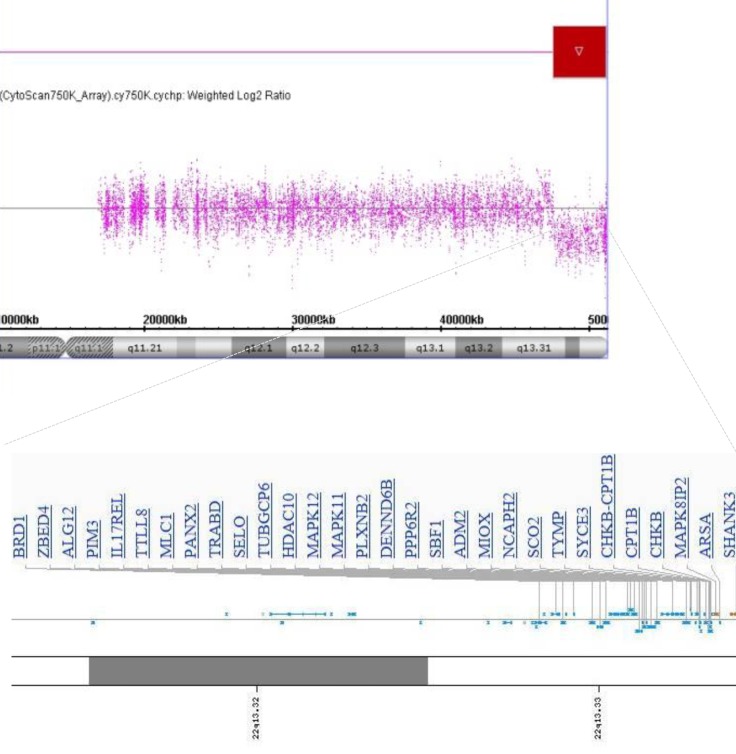
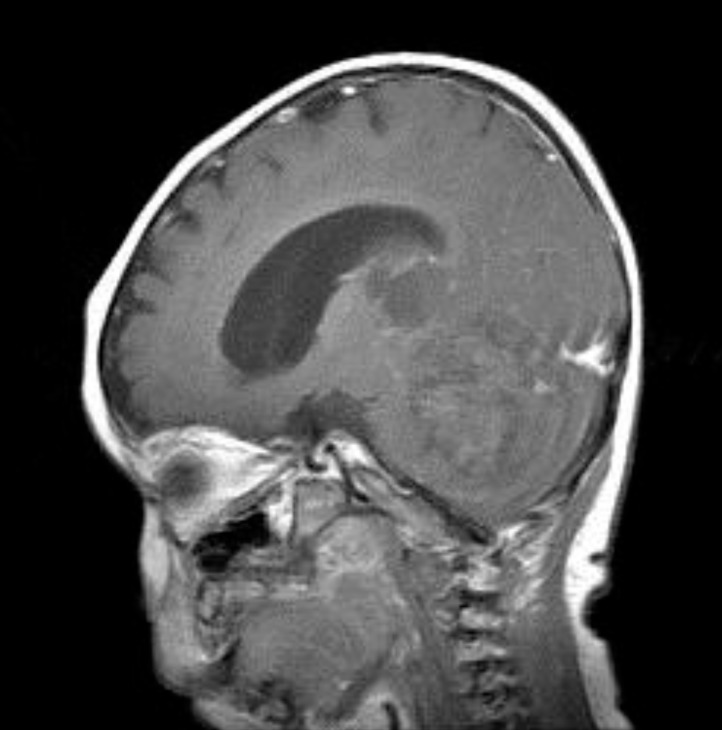
Here we present a 4 month-old-boy with 46,XY,r(22)(p13q13.3). High-resolution microarray analysis revealed a 3.5-Mb deletion at 22q13.31q13.33. At 11 months of age, an ATRT in the cerebellar vermis was detected after brain magnetic resonance imaging (MRI). To our knowledge, this is the first reported case of a patient with an ATRT and r(22) in Korea.
Case report
A 4-month-old boy was referred to Department of Pediatrics, Daegu Catholic University of Medicine Center because of delayed development. He was the first child of healthy, unrelated Korean parents without any known disorders in their family histories. The mother was 22 years old and the father was 27 years old at the patient's birth. The pregnancy was uneventful with no evidence of teratogenic agent exposure. The patient was born at 37 weeks of gestational age by uncomplicated spontaneous vaginal delivery, and he weighed 2,500 g. The physical examination revealed unremarkable findings, except generalized hypotonia with decreased deep tendon reflexes of both knee joint was found. Primitive reflexes could not be elicited. He could not maintain his head well a majority of the time. The patient weighed 7.4 kg (50th percentile), and was 65 cm tall (50th percentile). Results of the brain and abdominal ultrasound and the echocardiography revealed nonspecific findings. Cytogenetic analysis on peripheral blood lymphocytes revealed an r(22) in all analyzed cells, specifically 46,XY,r(22)(p13q13.3) (Fig. 1). To specify the breakpoint, high-resolution microarray analysis was performed. Upon analyses of the genomic DNA using an Affymetrix Cytoscan 750K array analysis (Santa Clara, CA, USA), a 3.5-Mb deletion at 22q13.31q13.33 was revealed (Fig. 2). However, the SMARCB1 (INI1/hSNF5) gene at 22q11.2, which lies proximal to the break point, was not deleted. Both parents had normal karyotypes. Thus, this chromosomal alteration of the proband was possibly de novo. At 11 months of age, the patient was brought to the Emergency Department with altered consciousness after falling out of his bed at home. Results of brain MRI revealed a 5.6-cm├Ś5.0-cm├Ś7.6-cm mass in the cerebellar vermis (Fig. 3). Results of the abdominal MRI revealed nonspecific findings. The operation was performed via transvermian approach by midline suboccipital craniotomy. The mass, extending the 4th ventricle and the quadrigeminal cistern, was resected. However, it's unfortunate that we obtained small biopsy specimens. On histopathologic examination, most tumor was exclusively composed of small cell component (Fig. 4A). The tumor cells, which had some cytoplasm and vesicular nuclei, were larger than cells of medulloblastoma. The typical rhabdoid tumor cells with eosinophilic cytoplasm were not appeared, and there were no organoid arrangements such as rosette or palisading pattern in the obtained tumor tissue. Immunohistochemical staining for expression of the INI1 protein, showed loss of nuclear expression in the tumor cells (Fig. 4B). After operation, the patient's condition deteriorated with status epilepticus. A ventriculoperitoneal shunt for hydrocephalus underwent in the referred hospital. The chemotherapy (vincristine, cisplatin, doxorubicin, and cyclophosphamide) has been done until now.
Discussion
Ring chromosomes are usually resulted from two terminal breaks in both chromosome arms, followed by fusion of the broken ends, or from the union of one broken chromosome end with the opposite telomere region, leading to the loss of genetic material. They can also be formed by fusion of subtelomeric sequences or telomere-telomere fusion with no deletion4). In a ring chromosome, the primary deletion associated with ring formation may be accompanied by a secondary loss or gain of material, and the instability in the ring chromosome contributes to the variable phenotypes observed4).
Although the phenotypic spectrum of r(22) is broad and can range from mild to severe2,7,8), carriers of r(22) present with most features of 22q13.3 deletion syndrome2,3). In both r(22) and 22q13.3 deletion syndrome, SHANK3 is suggested to be the most likely candidate gene influencing neurobehavioral features3). SHANK3 codes for a scaffolding protein that lies at the core of the postsynaptic density in glutamatergic synapses.
The 22q13.3 deletion syndrome (or Phelan-McDermid syndrome) typically results from the loss of the distal long arm of chromosome 22. This may result from simple deletions, an unbalanced translocation, or other structural rearrangements involving chromosome 229). Major clinical features include neonatal hypotonia, moderate to severe intellectual impairment, severe delayed or absent expressive language, normal to accelerated growth, autism-associated characteristics, and minor dysmorphic features9). Generally speaking, individuals with the 22q13.3 deletion syndrome have no life threatening organic malformations.
Carriers of r(22) may also present with the features of nurofibromatosis type 2-associated tumors such as vestibular schwannomas, multiple meningiomas, and neurofibromas. In these cases, the pathogenesis of these tumors was explained by the loss of both alleles of the NF2 gene (neurofibromin 2), a tumor suppressor gene located on chromosome 22q12.210,11). Although the NF2 gene was usually intact within the ring, the ring itself was prone to loss during somatic mitosis, and a pathogenic mutation at the NF2 gene on the remaining chromosome was thought to result in tumor development10,11).
SMARCB1 (INI1/hSNF5) is a tumor suppressor gene located on 22q11.2, proximal to the NF2 gene. In the majority of AT/RT cases, genetic alterations affecting the SMARCB1 (INI1/hSNF5) include homozygous deletions, heterozygous deletions as well as copy-number neutral loss of heterozygosity and mutations affecting each of all nine exons of SMARCB16). Immunohistochemistry using an antibody directed against SMARCB1 has evolved as a convenient first line diagnostic tool for the diagnosis of ATRT. This is especially relevant in small biopsy specimens, where rhabdoid tumor cells can be missed6).
In the past, CNS ATRTs were often misclassified as a medulloblastoma, primitive neuroectodermal tumors, or a different malignant brain tumor, because of their clinical, histological, and radiographic similarities6,12,13). However, they are separated from other embryonal tumors by the presence of rhabdoid cells and specific immunohistochemistry6,13).
The coexistence of a CNS ATRT in a child with an r(22) is rare. A previous case reported a CNS ATRT in a 4-year-old girl with r(22)14). In addition, a 2-year-old girl with 22q13.3 deletion syndrome and a CNS ATRT was reported15). In this case, although the array comparative genomic hybridization analysis of the patient's blood revealed only a de novo subtelomeric 7.2-Mb deletion of chromosome 22q13.2-q13.33, the results from the frozen tumor tissue demonstrated an acquired somatic frameshift mutation of the INI1 gene and the loss of the de novo germline 22q13 deleted chromosome15). The net effect was the homogeneous inactivation of the INI1 gene, leading to the development of the ATRT15).
Although we could not perform a genetic analysis of the tumor in our patient, we think that the ATRT may have resulted from the combined loss of the r(22) and a pathogenic INI1 mutation on the remaining chromosome 22. Therefore, monitoring for not only neurofibromatosis type 2-associated tumors, but also for ATRTs should be performed in all carriers with an r(22).
The familial transmission of r(22) from a normal woman to her clinically affected daughter was also reported. The benign r(22) in a parent may undergo further rearrangements not only during mitosis, but also during meiosis to produce an unbalanced chromosome associated with developmental abnormalities2). Therefore, both the gene deletion associated with ring formation, and the secondary genetic imbalance, should be considered.


 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


