


Introduction
3-methylcrotonyl-coenzyme A carboxylase (3MCC) deficiency is a rare defect of L-leucine metabolism1). The urinary excretion of 3-hydroxyisovaleric acid (3-HIVA) and 3-methylcrotonylglycine (3-MCG) is increased. 3MCC deficiency is one of the most common inborn errors of metabolism detected in neonatal screening programs (1/36,000 births) using tandem mass spectrometry2).
Although the manifestation of 3MCC deficiency is variable, most infants detected by tandem mass screening are asymptomatic and remain healthy during the follow-up, and a number of apparently asymptomatic mothers have been diagnosed with 3MCC deficiency through their infant's newborn screen3,4). Recently, there have been an increased number of cases of 3MCC deficiency in Korea after tandem mass spectrometry was introduced as the newborn screening test5,6). However, there is only one case report about maternal 3MCC deficiency in Korea7). We identified a case of asymptomatic maternal 3MCC deficiency detected by her son's tandem mass spectrometry.
Case report
A 14-day-old boy was referred to our Endocrinology and Metabolic Disease Clinic because of increased levels of 3-hydroxyisovaleryl carnitine on a neonatal screening test performed at one week of age.
The patient was born at gestational age 39+3 weeks by vaginal delivery. His birth weight was 3.2 kg, and he did not have immediate perinatal problems. There were no complaints of nausea, vomiting, or poor oral feeding. He did not show tremor, seizures or any other neurologic symptoms. He is the first baby of his parents. There was no specific medical history in his family.
He was not ill looking. His height was 54 cm (0.26 standard deviation score), weight was 3.8 kg (-1.22 standard deviation score), and head circumference was 37 cm (50-75 percentile). His blood pressure was 80/49 mmHg, pulse rate was 142 beats/min, respiratory rate was 31 breaths/min, and body temperature was 36.6℃. The physical exam was unremarkable. He was alert and showed normal findings on the neurologic examination, and his muscle tone was not hypotonic.
Leucine-free formula feeding and carnitine (100 mg/kg/day) supplementation was started because we suspected he had a 3MCC deficiency.
The laboratory studies showed a normal complete blood count, normal electrolytes, and normal renal and liver function test results. In addition, there was no acidosis on the venous blood gas analysis, and his ammonia level was also within the normal range.
Metabolic investigations showed normal serum amino acids and normal concentrations of free and total carnitine. 3-MCG and isovaleric acid were not detected in the urine organic acid analysis.
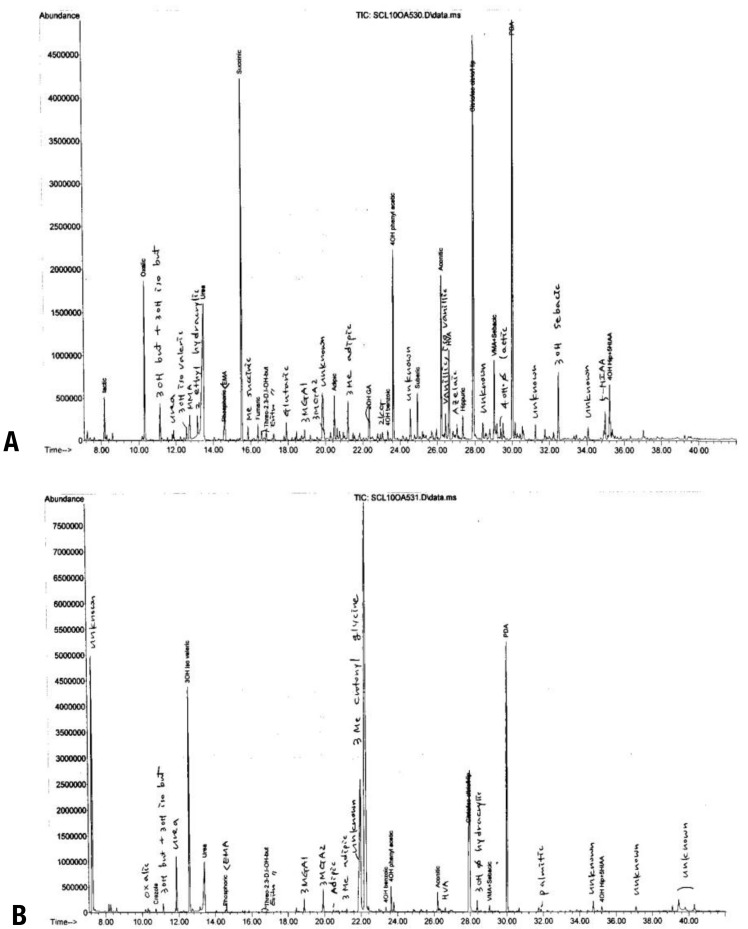
We then suspected maternal 3MCC deficiency, and a urine organic acid analysis of the patient's mother was performed. The 3-MCG level was increased (227.98 mmol/mol Cr, control: not detectable) in her urine (Fig. 1). She was diagnosed as having maternal 3MCC deficiency. She was asymptomatic until diagnosed as having 3MCC deficiency. There were no abnormal findings on her physical examination, and she was not lethargic and had no specific symptoms of acute illness according to her past medical history. We did not perform an exact IQ test, but her intellect appeared normal. We recommended further evaluation such as a plasma amino acid analysis, enzyme activity test and genetic study. However, she refused further investigation and gene analysis because she was asymptomatic.
After maternal 3MCC deficiency was diagnosed, the leucine-free formula and carnitine supplementation for her baby was stopped, and breast milk feeding was started.
After 3 months, the mother and her baby were doing well, and they had no symptoms associated with 3MCC.
Discussion
3MCC deficiency is an abnormality of leucine metabolism1). It is induced by mutations in the MCCC1 and MCCC2 genes encoding the α and β subunits, respectively8).
3-HIVA and 3-MCG are increased in the urine of 3MCC deficiency subjects. Additionally, 3-hydroxyisovalerylcarnitine is also high in the blood and urine, and it is detected by the neonatal screening test. Carnitine deficiency can also develop in many 3MCC-deficient patients1).
Signs/symptoms previously described in 3MCC deficiency include poor feeding, vomiting, hypotonia, seizures, failure to thrive, developmental delay, and Hypoglycemia1). However, the neonatal screening test using tandem mass spectrometry, which has become quite prevalent recently, has revealed that the clinical picture of MCC deficiency is heterogeneous and often highly variable, even within the same family4,9,10). Some patients usually have no symptoms but do show symptoms such as vomiting, involuntary movements, seizures, coma and apnea associated with metabolic acidosis, hyperglycemia and mild hyperammonemia only during concurrent infections or illnesses10,11,12). The majority of children diagnosed by the neonatal screening test have been reported to have remained asymptomatic so far6,13,14).
3MCC deficiency is inherited as an autosomal recessive trait. Therefore, the importance of screening for the defect in the asymptomatic siblings of affected patients has been established15). Otherwise, we are usually not concerned with the parents of the proband. However, several asymptomatic 3MCC-deficient mothers have been identified only by abnormal results in the neonatal screening test from their unaffected babies4,13), which was observed in our case. The abnormal metabolites found in the healthy infants originated from the mothers, who are unrecognized 3MCC-deficient patients, via placental transfer4). Therefore, metabolic analysis of the mother in addition to screening positive infants is recommended. The mother should have a urine organic acid analysis and a plasma acylcarnitine profile15).
There are few reports about maternal 3MCC deficiency3,4,7,9). Most affected mothers identified through their infant's abnormal screening results were asymptomatic despite having consumed an unrestricted diet throughout their life. However, one of 4 affected mothers reported significant emesis with minor illness or after high protein meals3). An additional 2 of 4 affected mothers reported by Gibson et al.4) had mild symptoms such as fatigue, myopathy, weakness and elevated liver enzymes. According to the report by Grunert et al.9), 1 of 8 maternal 3MCC-deficient patients had symptoms such as several metabolic crises with hypoglycemia during febrile illnesses, metabolic stroke, cardiomyopathy, and paresthesias. In a Korean case report7), the affected mother was asymptomatic, which is the same as our case.
Supplementation with oral L-carnitine and a leucine-restricted diet is the treatment for 3MCC deficiency, but the efficacy of these approaches is unproven15). Moreover, there is consensus that leucine-restricted diets are not indicated for asymptomatic mothers and that the decision regarding restricted diets in symptomatic mothers should be individualized15). Because the mother of our case was asymptomatic, we did not restrict her diet or supply carnitine.
In our case, the confirmatory enzyme analysis and genetic analysis of the mother were not performed. Although, confirming the diagnosis based on the enzyme assay whenever possible is preferable, there is consensus that 3MCC deficiency can sometimes be diagnosed based on metabolite levels alone without the enzyme analysis, depending on the specific metabolites and degree of elevation15).
Moreover, the genotype does not appear to be predictive of the phenotype or metabolic risk13). Thus, DNA analysis is not yet helpful in the management of affected patients, but it can assist with the confirmation of questionable cases15).
We identified asymptomatic 3MCC deficiency in an adult detected via an abnormal neonatal screening test result of her unaffected baby. Although maternal 3MCC deficiency is uncommon and the investigation of asymptomatic mothers is difficult, we can confirm that investigating neonatal screening-positive infants and their mothers simultaneously is important.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


