


Introduction
Griscelli syndrome type 2 (GS2) is a rare autosomal recessive disorder, characterised by partial albinism, immunodeficiency, organomegaly and accelerated phases1). During the accelerated phases, haemophagocytosis and pancytopenia occur which may be accompanied by neurological deterioration1,2,3,4). Isolated neurological deterioration without hematological involvement has been rarely described5,6,7,8). We describe two brothers who had GS2 with varying clinical manifestations. The elder boy had an isolated neurological presentation, while the younger brother had hematological features without neurological manifestations. A novel mutation in the RAB27A gene was detected, genetically confirming the diagnosis.
Case report
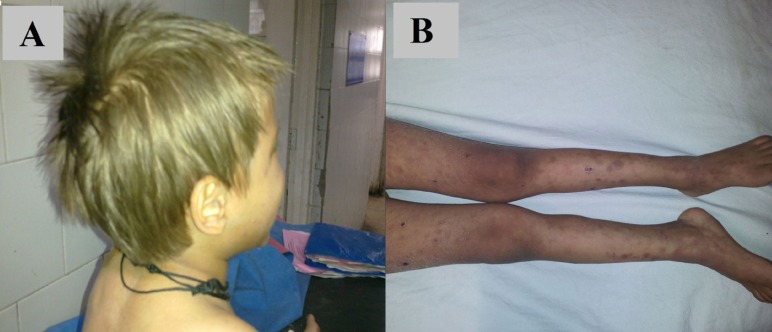
A 5-year-old boy, born out of nonconsanguineous marriage, second in birth order, presented with high grade intermittent fever and a rash over the lower limbs, for 3 months. His elder brother had expired 6 months back at 7 years of age due to an undiagnosed medical condition. On examination, the patient, of average built and nutrition, was febrile, with a heart rate of 142/min, respiratory rate of 22/min, blood pressure of 102/64 mmHg, severe pallor, hepatosplenomegaly (liver 2 cm below right costal margin and spleen 3.5 cm below left costal margin) and a soft ejection systolic murmur. He had a generalized hypopigmented skin with golden-brown hair (Fig. 1A). Both lower limbs showed multiple, well-defined, tender, erythematous to hyperpigmented, nodular lesions, extending from midthighs to feet, some having scab formation (Fig. 1B). The parents revealed that their deceased elder son had similar hypopigmented skin, though no other family member was affected.
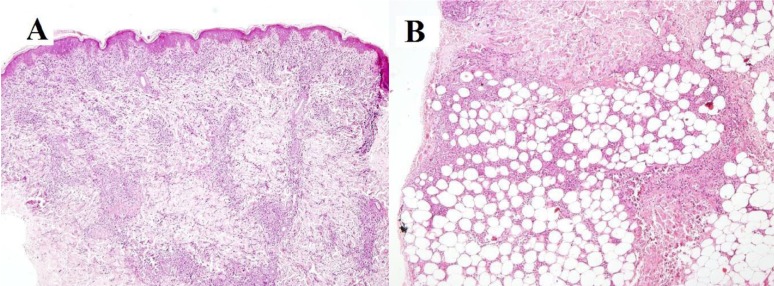
Investigations at admission showed hemoglobin, 4.1 g/dL; total leucocyte count, 2,500/mm3; lymphocyte, 73.7%; neutrophils, 18.6%; platelet, 33,000/mm3; erythrocyte sedimentation rate, 11 mm (1st hour); C-reactive protein, 21 mg/L. The peripheral blood smear showed normocytic, normochromic red blood cells with mild anisocytosis, with a corrected reticulocyte count of 3.2%. The bone marrow aspiration smears showed erythroid hyperplasia, predominantly normoblastic, with normal maturation of myeloid series, no megakaryocytes and no abnormal cells or parasite. Neither the peripheral blood smear nor the bone marrow aspiration smears showed large granules in neutrophils, ruling out the possibility of Chediak Higashi syndrome. The results of kidney and liver function tests, serum electrolytes, uric acid, chest skiagram and Mantoux test were within normal limits. The blood and urine cultures were sterile. Investigations to rule out common infections like Widal test, rapid malaria antigen test, rK 39 antigen, Dengue serology, and Weil Felix test did not reveal any abnormalities. Also, hepatitis B surface antigen and human immunodeficiency virus serology were negative and his immunoglobin levels were within normal range. Serum ferritin was 1,334 ng/mL. The antinuclear antibody (ANA) showed speckled positivity (1+), anti-dsDNA was borderline positive at 50 IU/mL, RA factor being negative. Ultrasound of the abdomen was normal except for enlarged spleen (13.5 cm) and enlarged liver (15 cm). Skin biopsy taken from the lower limb lesions was suggestive of erythema nodosum (EN) (Fig. 2A, B).
The patient was treated with antipyretics, blood product transfusions and intravenous ceftriaxone. However, the patient's condition deteriorated with worsening pancytopenia, increasing size of liver and spleen and aggravation of the rash. Considering the weakly positive ANA and presence of EN, a possibility of a connective tissue disorder was considered and an empirical trial of oral prednisolone at 2 mg/kg/day was started.
While investigations for this patient were in progress, efforts were made to trace the medical records regarding the illness of his elder sibling. This child had presented with fever with raised intracranial tension, rapidly progressive spastic quadriparesis, and papilledema. He had no organomegaly and his blood counts were normal. An initial computed tomography (CT) scan of the brain showed obstructive hydrocephalus. The cerebrospinal fluid (CSF) examination showed 25 cells, (100% lymphocytes) with protein 112 mg/dL and glucose 80 mg/dL. Based on the CT and CSF findings, he was started on antitubercular treatment with steroids. However he continued to worsen and a magnetic resonance imaging (MRI) of the brain was obtained which showed extensive signal abnormalities in the cerebellar white matter and cortex, with inhomogeneous signal abnormalities in the thalamus, basal nuclei, internal capsule, and cerebral white matter. There was evidence of obstructive hydrocephalus with impending tonsillar herniation. There was a dot-like contrast enhancement in the cerebellum. After an expert review of the MRI, a possibility of vascular-perivascular disorders like malignancies or hemophagocytic lymphohistiocytosis (HLH) was considered. Bone marrow examination was normal. The patient underwent a ventriculoperitoneal shunt surgery along with a brain biopsy from the right parietal region. However, a very small volume of material was available from the brain biopsy which showed nonspecific lymphocytic proliferation. The patient could not be investigated further, as he continued to deteriorate, and died two weeks later.
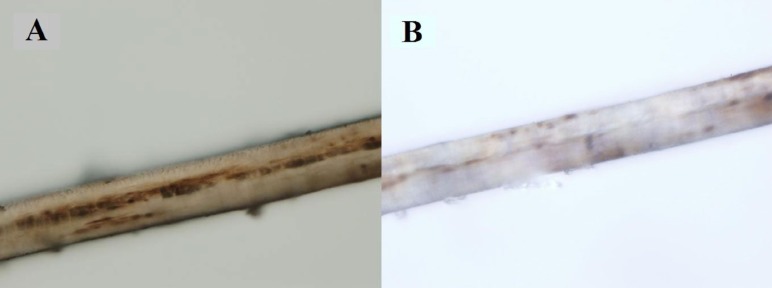
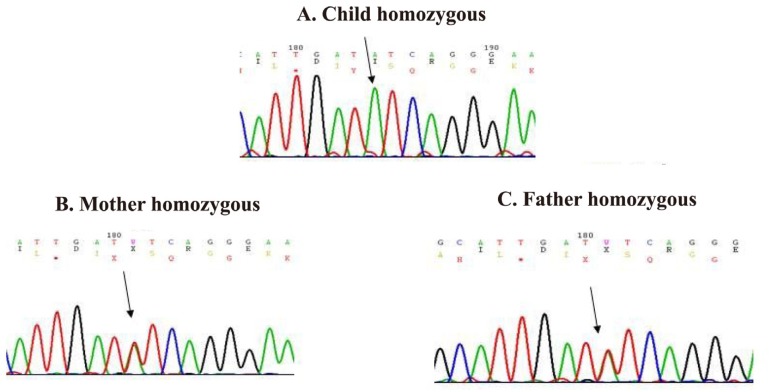
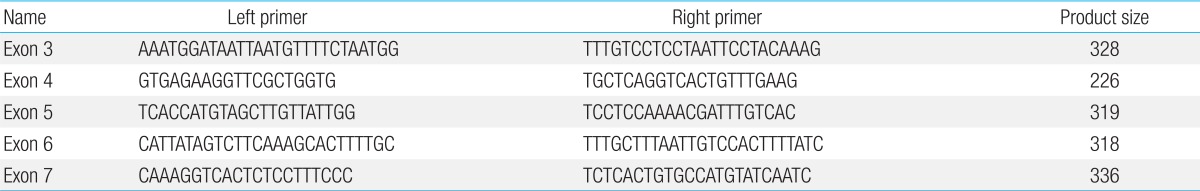
After obtaining information about the elder sibling's illness, it was inferred that both the siblings were suffering from a common disorder of hypopigmentation with potentially fatal phases with two varied manifestations. A possibility of GS2 in accelerated phase was considered. Subsequently, a hair shaft examination of the younger sibling was done, which revealed large sized melanin granules, distributed irregularly along the length of the hair shaft mainly near the medulla. (Fig. 3A). Under polarized light microscopy, the hair appeared monotonously white (Fig. 3B). A genetic analysis was done to detect mutation in RAB27A gene. Genomic DNA was extracted from peripheral blood sample and sequencing of RAB27A gene was done. The sequence of the pairs of primers used to amplify these regions is shown in Table 1. Mutation analysis of RAB27A gene revealed a novel homozygous mutation in RAB27A gene on chromosome 15 (exon 3) showing a single-base substitution (c.136T>A p.F46I), leading to an amino acid change from phenylalanine (TTC) to isoleucine (ATC). This mutation is damaging by Polyphen. Parents were found to be heterozygous for the same mutation (Fig. 4A-C).
Hence the patient was diagnosed as a case of GS2 with HLH; his elder sibling was retrospectively diagnosed to have GS2 with cerebral lymphohistiocytic infiltration. The patient achieved normalization of blood counts on oral prednisolone. The parents were counseled about the disease, and possible need for bone marrow transplantation.
Discussion
Griscelli syndrome was first described in 19789). Mutations in MYO5A, MLPH, and RAB27A genes are responsible for the differing manifestations of type 1, 2, and 3 respectively. Partial albinism is common to all the 3 types with additional features of a primary neurological disorder seen in type 1 and features of variable immunodeficiency being seen in type 21,7,10,11).
The primary immunodeficiency seen in GS2 is due to an impairment of T cell and natural killer cytotoxic activity, resulting in susceptibility to repeated infections, ultimately reaching a fatal accelerated phase characterized by HLH4,12). This phase usually presents with periods of fever, hepatosplenomegaly and pancytopenia13,14). In addition, a wide spectrum of neurological features may be seen due to cellular infiltration of the brain1,2,3,4). A little over 100 cases of GS2 have been reported so far in medical literature, half of which have had their mutation sequencing done1,13). Most of the cases were from Turkey and Iran. The mean age of development of symptoms of HLH ranged from 2 months to 13 years13,15). Reports from India have been scarce15).
The siblings with GS2 described by us, not only carried a novel mutation in RAB27A gene, but also presented in an unusual manner. The elder boy manifested a purely neurological picture with development of hydrocephalus and rapid deterioration, without any hematological abnormalities or hepatosplenomegaly, quite unsuspecting of GS2. Such cases of GS2 with isolated neurological involvement in the absence of hematological manifestations of HLH have rarely been reported5,6,7,8). His brother had a hematological presentation with fever, pancytopenia and hepatosplenomegaly. The unusual finding in this child was the presence of EN, which has not been described with Griscelli syndrome before; rather, this finding shifted the focus to other conditions known to be associated with EN. It is a nonspecific cutaneous reaction pattern to a variety of antigens, with many immune-mediated mechanisms implicated. Most direct and indirect evidence supports the involvement of a type IV delayed hypersensitivity response to numerous antigens16). The abnormal T-cell function in Griscelli syndrome may have been a contributing factor in this patient.
The diagnosis of the elder sibling could only be made retrospectively after the genetic analysis of the younger sibling and his parents. It is also interesting to note that 2 siblings with the same genetic mutation can present with two entirely diverse clinical pictures.
The prognosis of GS2 is poor, with death in early childhood due to HLH and its complications. Hematopoietic stem cell transplantation is the only curative treatment for these patients14,17).
Few case reports similar to ours have been reported in which diagnosis of GS2 could only be made in the second sibling, the earlier sibling having died abruptly1,7,15). This emphasizes the need for greater awareness of this potentially fatal entity which can have heterogenous clinical manifestations. Earlier molecular diagnosis can help prompt initiation of treatment of the affected child and genetic counseling and antenatal diagnosis for future conceptions.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


