


Introduction
Growth is a very important feature of development of human in which several environmental and genetic factors involved1). Short stature is defined as a height more than 2 standard deviations (SDs) below the mean for age and sex, comparing to the national height standards or one that is below the third percentile for the age and gender in a population2). About 2%–3% of the children in the world have short stature3). Eighty percent (80%) of the cases have no history of small for gestational age, growth hormone (GH) deficiency, or other pathologies, therefore, this group of patients defined as idiopathic short stature (ISS)2). A small proportion of genes are concerning to short stature4). Short stature homeobox (SHOX gene MIM 312865) is one of the major genetic contributors to human growth. SHOX mutations can cause Leri Weill dyschondrosteosis and Langer mesomelic dysplasia. Moreover, Short stature in Turner syndrome is the result of SHOX gene haploinsufficiency5). In 1997, Rao et al.4) were the first to describe SHOX gene mutations in individuals with ISS (ISS MIM 300582). SHOX gene is found in the pseudoautosomal region 1 of the short arms of both human sex chromosomes at the position Xp22.3-Yp11.3 and consists of six exons, the first is noncoding and the others coding4). Several clinical and molecular studies identified the SHOX gene as the cause of the short stature phenotype6). Intragenic mutations are described in 2%–15% of cases7). Previously, it has been proven that the conserved noncoding elements downstream of the SHOX gene regulate gene function8). These conserved elements present a 250-kb downstream regulatory domain that may be deleted in patients with ISS and in other SHOX gene defects with a frequency of 22%3).
Materials and methods
Prospective study of 105 children (57 female and 48 male childrens) were diagnosed clinically as having ISS and screened for mutations of the SHOX gene. Their ages ranged from 2 to 18 years with mean of 11 years. They were enrolled between September 2013 and December 2015 and referred by the pediatric endocrinology outpatient services of Armed Forces Hospital, Taif region, KSA, under the prior approval in accordance with the principles of the Regional Research and Ethics Committee of Hospitals (IRB, H-02-T-001).
Patients with normal thyroid function tests, normal karyotype in females, negative celiac screening, negative chronic organic/psychological or syndromic diseases, normal 2 GH provocation test with peak value ≥10 µg/L (to exclude GH deficiency)12) and lack of clear signs of chondrodysplasias were entered this study. These inclusive criteria were chosen according to the proposed consensus on the definition of ISS2), as it is a diagnosis of exclusion. For every patient; measurement of weight (kg), standing height (cm), arm span (cm) (which is the length from one end of an individual's arms (measured at the fingertips) to the other when raised parallel to the ground at shoulder height at a 90°13), sitting height (cm) (which is the distance from the highest point on the head to the base sitting surface)13) and calculation of subischeal leg length (cm) (which is the difference between height and sitting height)13) was performed. Also, paternal height (cm) and maternal height (cm) were measured for every patient, to calculate the mid parental height (for boys: [paternal height+{maternal height+13 cm}]/2, for girls: [maternal height+{paternal height–13 cm}]/2). X-ray of the left hand was evaluated for bone age and peripheral blood samples were collected from the patients for DNA extraction.
Genomic DNA was extracted from peripheral blood leucocytes using an extraction kit (Thermo Fisher, Waltham, MA, USA) and used for polymerase chain reaction (PCR) amplification. The entire regions of exons of the SHOX gene were amplified by PCR using the newly designed specific primers. The PCR products were confirmed by agarose gel electrophoresis.
Direct sequencing of purified products using the same primers (Table 1) was performed on both strands using the Big Dye Terminator Cycle Sequencing Ready Reaction Kit on a 3130 Genetic Analyzer (Applied Biosystems, Waltham, MA, USA) and the raw sequencing results were collected using the Data collection and analyzed using sequencing analysis software version 3.1 (Life Technologies, Grand Island, NY, USA). Seqscape software version 2.7 (Applied Biosystems) was used for base-calling and mutation detection. All SHOX variants reported in this study were submitted to the Human Variation Database of the National Center for Biotechnology Information.
Results
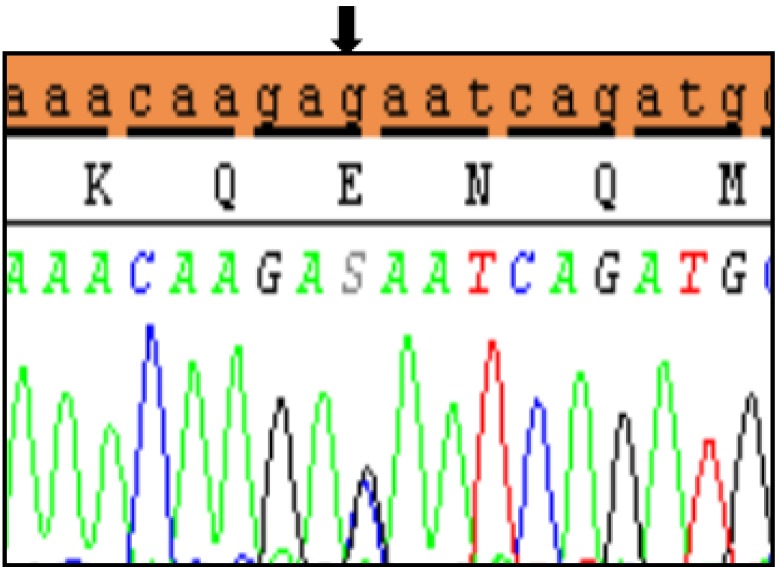
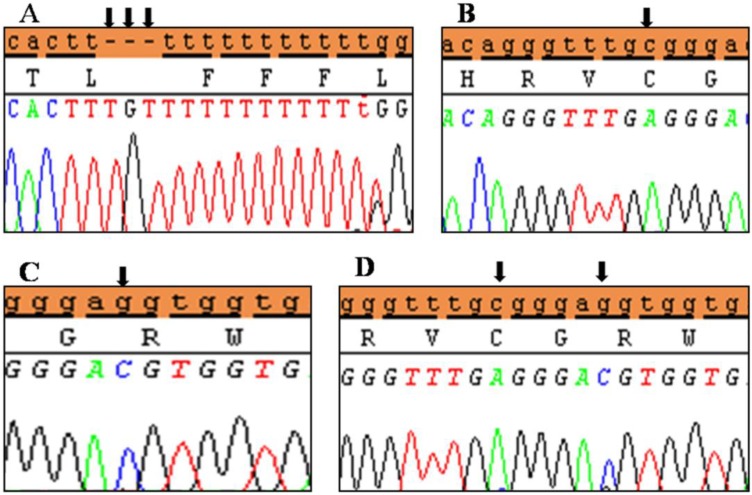
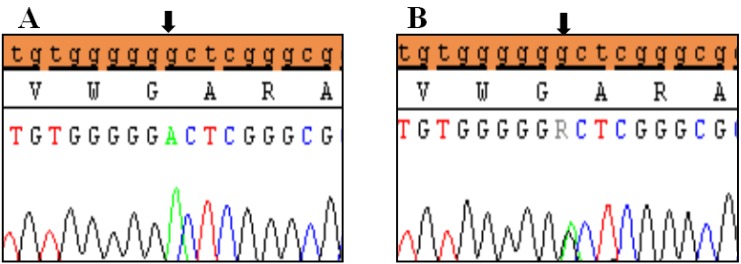
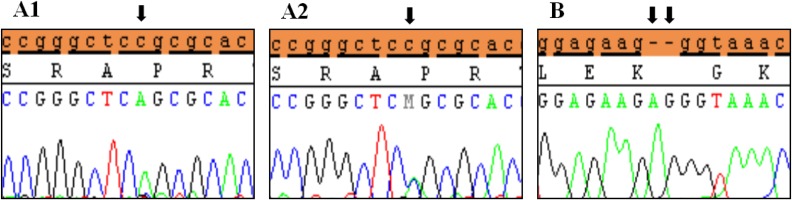
Clinical characteristics and, mean of all anthropometric measures of ISS patients included in the study (both mutated and nonmutated) are shown in Table 2. Of all ISS children analyzed 17 girls and 13 boys have variants indicate that females more affected than males with ratio of 57% females and 43% males in ISS patients with variants. These results revealed a 28% frequency of alterations to the SHOX gene in Saudi Arabia children with ISS. Anthropometry of ISS patients with SHOX variants and those without revealed that there are no big differences between mean height, mean weight, arm span, sitting height and subischeal leg length in both groups within males gender while big differences were noted in these parameters in females, suggesting that females more to be affected by SHOX variation. Direct DNA sequencing of purified PCR products resulted in overall with 1 mutation and 6 polymorphic variants. Four variants were previously reported in the SHOX database and the others have not been described and defined as novel. These mutation and variants are located in exons 1, 2, 4 and 6, while, no variants were detected in exons 3 and 5. Interestingly, we found a missense mutation (c.528G>C, p. E176D) in the DNA homeodomain binding site of exon-4 at the position 528. This mutation was detected in 1 girl patient aged 5.5 years as a heterozygous form (Fig. 1). According to the PolyPhen-2 website program, this mutation is predicted to be probably damaging and considered as pathogenic one. Three polymorphic variants identified in noncoding region of exon-1. First variant is a novel and found as insertion of 3 nucleotides between the position c.-646_-645insTGT (Fig. 2A). The second and third variants were reported previously14) and detected in positions c.-512C>A and c.-507G>C, respectively, as a homozygous genotype forms (Fig. 2B, C). Interestingly, 6 patients were carried both variants (c.512C>A and c.-507G>C) as a compound homozygous (Fig. 2D). Next, 1 polymorphic variant was identified in 5'UTR (untranslated region) of exon-2, in position c.-372 G>A as homozygous form in five patients and heterozygous form in 9 cases (Fig. 3A, B). Two novel polymorphic variants were identified in exon-6a. First one was detected in position c.*41C>A in the 6a- 3'UTR as homozygous in 1 patient and heterozygous genotype forms in the other one (Fig. 4A1, A2). Second variant was found as an insertion variant detected in position c.*284_285insAG in 6a- 3'UTR (Fig. 4B). Table 3 summarized the phenotypically characteristics and polymorphic variants frequencies that found in this study. There is no significant differences in the phenotypic parameters between ISS patients that having DNA variations and those without DNA variations.
Discussion
Longitudinal growth is a complicated process that is controlled by genetic factors and regulated by both permissive and regulatory factors15). SHOX gene belongs to the transcriptional regulators family that has an essential role in development16). SHOX gene variants expected to be responsible for 2% of patients with ISS17) that has increased to 23% by time due to better clinical attention to such gene18). Our study of 105 children with ISS showed 30 patients (28%) of the children had at least 1 variant that explained their short stature. The higher frequency in our study when compared to published studies that reported this association with a very broad frequency range varying from 1.5% to 15%19), which may be explained by ascertainment bias or by a true increased frequency of SHOX variants as a result of high rate of consanguineous marriage in Saudi Arabia as reported by el-Hazmi et al.20).
Comparison of mutation prevalence in prior studies is difficult due to varying definitions of ISS, the inclusion of patients with skeletal abnormalities, different mutational analyses (single-strand conformational polymorphism vs. direct sequencing), differences in the regions of the SHOX gene analyzed, and population diversity. Our variants rate is higher than the mutation range as previously reported by Rappold et al.21), of a large cohort of 1,534 patients with ISS, a mutational frequency of 2.2% was documented. Huber et al.6) found a frequency of 15% in 84 patients, Sandoval et al.22) found a frequency of 8.1% in 62 ISS Colombian patients, while Chen et al.3) found a frequency of 22%. Interestingly, in this study, most of our male patients present at school age, while females presented earlier and shorter. This result was in agreement with Binder, who noted that although the penetrance of SHOX deficiency is high, but its clinical expression is very variable seemly more marked with age23). Characteristic signs are more frequent and more severe in girls, a finding which may be explained by the presence of higher estrogen levels in females24). Previous studies showed several mutations in SHOX gene in ISS patients in some countries22,25).
Only few clinical studies related to pattern, diagnosis and frequency of short stature in Saudi Arabia children were done9,10,11). The molecular genetics techniques allowed the opportunity to improve the diagnoses of disease especially in the field of endocrinology science. With respect to variants found in the present study and those previously reported in literature, 2 polymorphic variants in noncoding region of exon-1 (c.-507G>C, c.-512C>A) and one polymorphic variant was identified in 5'UTR of exon-2 (c.-372G>A) were already reported14).
According to the hypothesis of the ability of regulatory elements to modulate gene expression26), and as reported previously, the 5′ UTR has an important role in posttranscriptional regulatory pathways that control mRNA localization, stability and translation efficiency27), the high frequency of variants that identified in noncoding regions of exons 1 and 2 as a promoter region could be affecting the expression of SHOX protein.
Interestingly, we detect a pathogenic mutation in the homeodomain DNA binding site of SHOX gene in exon 4 (c. 528G>C). This homeodomain play important role in DNA binding, protein transactivation, cell cycle and growth regulation28). Clement-Jones et al.16) stated that the homeodomain proteins have an important physiological functions in regulating the embryonic development in vertebrates, suggesting that the mutations in SHOX gene especially in homeodomain region may lead to developmental abnormalities. Other studies revealed the functional consequences for the mutants R168W, L132V, A170P and R173H that found in homeodomain region of SHOX gene with respect to DNA binding, dimerization or nuclear translocation of the SHOX protein 29,30).
In this study, 3 novel variants were found, one in exon 1 and 2 variants in the 3' UTR of exon-6a region. Thomas et al.31) reported that there is a high phenotypic variability related to gain or loss of SHOX gene sequences and their regulatory regions. The analysis of a region located at 3' of the gene has been included in the screening of short stature, since some studies suggested the presence of distal regulatory elements of SHOX transcription in this region 8,32). Recently, Solc et al.14) documented the common variants in the coding and noncoding sequences including 5' region of SHOX gene.
Several studies, including the present one, employed similar measures criteria to identify ISS33,34). No significant differences were found regarding the SD of height, mid parental height, arm span, sitting height, subischeal leg length, and age within ISS patients. This can be explained by the suggestion of Rosilio et al.35) that anomalies in SHOX enhancer regions (i.e., noncoding region) seemed to be linked to a milder phenotype. This approves the fact that, the correlation between phenotype-genotype in patients with ISS is hard to detect. It might have been expected that children with variants in SHOX gene would had more severe short stature than those without variants in SHOX gene, however, we found no remarkable differences in the degree of short stature between those with or without identifiable SHOX gene polymorphic variants. Thus, there is no clinical parameter that serves to distinguish ISS patients with SHOX mutation from that idiopathic nonendocrine short stature without SHOX mutation.
This results was in agreement with finding of Rappold et al.30) as they reported that no significant difference of the mutations frequency was detected when comparing males and females in ISS pediatric patients. Similarly, L05101 deletion frequency between men and women in ISS patients are not significantly difference14).
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


