

Introduction
Kabuki syndrome (KS; OMIM 147920) was first described in 1981 by Niikawa et al.1) and Kuroki et al.2). It has an estimated incidence of 1 in 32,0003) and approximately 400 cases have been reported worldwide. In Korea, in 1991, Park et al.4) first reported KS patient and from 1991 to date, about 10 KS patients5-8) have been reported. However, none of these demonstrated causative genetic abnormalities. Consensus of clinical diagnostic criteria for KS have not been established. The main clinical characteristics of KS include five cardinal manifestations, as defined by Niikawa et al.3): typical dysmorphic facial features, skeletal anomalies including spinal column abnormalities and brachydactyly, dermatoglyphic abnormalities such as persistent fetal finger pads, mild to moderate intellectual disability and postnatal growth deficiency. The typical facial characteristics observed in KS include elongated palpebral fissures with eversion of the lateral third of the lower eyelid, arched or nicked eyebrows with the lateral third displaying sparseness or notching, short columella with depressed nasal tip and large, and prominent or upped ears.
A gene causing KS was identified through whole exome sequencing by Ng et al.9), who reported mutations in MLL2 in 66% of 53 patients with KS. In five published series, mutations in MLL2 were found in 56-76% of KS patients9-13). The recent search for additional causative genes revealed KDM6A deletion in three KS patients among 22 MLL2 mutation-negative patients14), and none KS patients among 120 MLL2 mutation-negative patients15). However, until now, the major cause of KS is known to be MLL2 gene mutations. In this report, we describe a case of a novel MLL2 gene mutation in a KS patient with typical distinctive facial features.
Case report
The male patient was born at 40 weeks gestation by spontaneous vaginal delivery to a 27-year-old G0P0 mother and a 29-year-old father. The parents were nonconsanguinous and healthy. The pregnancy and labor were uncomplicated. There was no history of fever, rash, spotting, tobacco, alcohol, illicit drug use, or X-ray exposure during pregnancy. At birth, the patient had microcephaly (<10th percentile) but no anatomical anomalies. He was admitted to a neonatal intensive care unit for 2 weeks due to transient respiratory difficulty. At 18 months of age, he first visited our clinic due to developmental delay. His height, weight, and head circumference were 77.4 cm (10-25th percentile), 11 kg (25-50 th percentile), and 43.4 cm (5-10th percentile), respectively. He could stand up with support at 15 months of age and walked alone at 25 months of age. His speech was delayed and at 27 months of age, he first said "mama". He showed long palpebral fissures with lateral eversion of the lower eyelid, long eyelashes, arched eyebrows with lateral thinning, and a depressed nasal tip. He had brachydactyly and prominent finger pads of bilateral third and fourth fingers. His intelligence quotient was estimated at 53 by Korean-Wechsler Preschool and Primary Scale of Intelligence, which meant moderate mental retardation (0.05th percentile). Spine X-ray showed butterfly vertebra at the 3rd thoracic spine, and echocardiography showed no cardiac anomalies except bicuspid aortic valve. Neurologic examinations showed no abnormal findings. He suffered from recurrent otitis media. At 11 years of age, his height, weight, and head circumference were 131.7 cm (<3rd percentile), 48.6 kg (75-90th percentile), and 55.5 cm (95-97th percentile), respectively. His chromosome was normal, 46, XY, and other laboratory findings including immunoglobulin, insulin, fasting glucose, cholesterol and thyroid hormone (TSH and free T4) were normal.
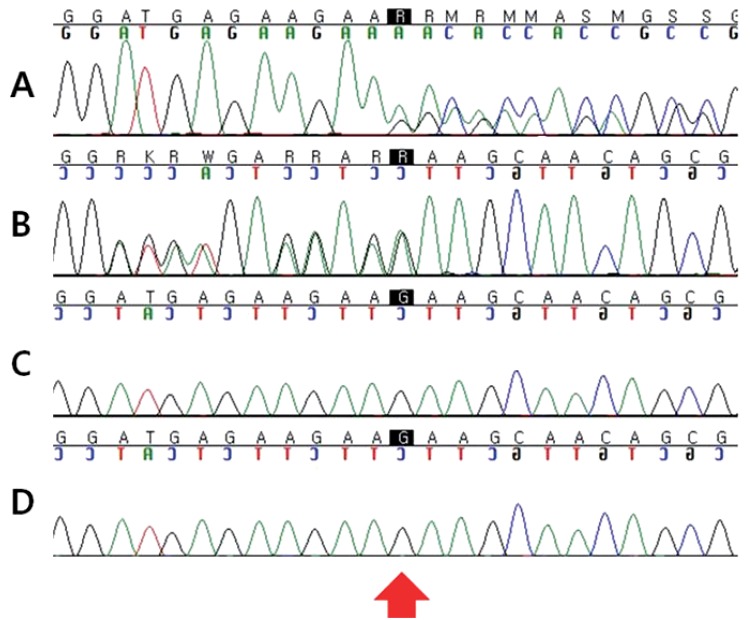
As our patient had typical facial features, skeletal anomalies and postnatal growth deficiency such as height 2 standard deviations, we diagnosed him as KS by clinical findings. After informed consent had been obtained, we performed molecular genetic testing for the MLL2 gene. A novel heterozygous MLL2 mutation (c.5256_5257delGA;p.Lys1753Alafs*34) was identified. However, no genetic mutations were detected in both parents (Fig. 1).
Discussion
MLL2 is 36.3 kb in size and comprises 54 exons. It is located at chromosome 12q13 and encodes a histone methyltransferase that regulates the transcription of a diverse set of genes involved in embryogenesis and development16-18). Recently, de novo partial or complete deletion of the Xp11.23 located gene, lysine demethylase 6A, KDM6A, has been identified in three KS patients, confirming the genetic heterogeneity of the disease14,15). Like MLL2, KDM6A plays a role in embryogenesis and development. KDM6A and MLL2 act together in the epigenetic control of transcriptionally-active chromatin.
Taking several studies10,11,13) together, the majority of MLL2 mutations were nonsense and frameshift. Hannibal et al.13). found MLL2 mutations in 74% of 110 KS patients, including 37 nonsense, 22 frameshifts, 16 missense, 3 in-frame deletions/duplications and 3 splice-site mutations; there were 10 sites at which recurrent mutations were observed. Li et al.11) found MLL2 mutations in 56% of 34 KS patients and 1 recurrent missense mutation (c.15461G>A). Micale et al.12) found MLL2 mutations in 73% of 62 KS patients and 3 recurrent nonsense, 3 frame shift mutations and 1 splice site variant. Missense mutations have been reported but are relatively infrequent10,11,13). In prior studies, mutations wer e distributed throughout the gene, however, there appeared to be some exons in which mutations were identified with considerably higher frequency such as exons 39 and 4813,19). It will be important in future studies to define that there might be mutational hotspot of particular relevance.
Comparison of clinical characteristics of MLL2 mutation-negative versus MLL2 mutation-positive cases allows us to explore both the relationship between MLL2 genotype and phenotype. Overall, some authors found growth retardation and distinctive facial features more frequently in MLL2 mutation-positive cases. However many studies found no significant different characteristics between MLL2 mutation-positive cases from MLL2 mutation-negative cases, except renal anomalies (single fused kidneys, ureteropelvic junction obstruction, duplication of the collecting system, and hydronephrosis) and short stature11,13,19,20). Renal anomalies and short stature were more common in MLL2 mutation-positive cases. In comparison with other MLL2 positive patients, our patient has short stature (<3rd percentile), however, he is overweighted (75-90th percentile) and he has macrocephaly (95-97th percentile).
Diagnosis of KS is mainly clinical, based on a combination of distinctive dysmorphic face, intellectual disability, and multiple congenital abnormalities. There are KS-like syndromes such as X-linked mental-retardation syndrome by defects of histone methylation which shows intellectual disability and malformations (e.g., cleft lip/palate). The histone methylases and histone demethylases have been implicated in multipleanomaly syndromes. In clinical practice, having the molecular confirmation of the clinical and biochemical diagnosis helps differential diagnosis. In addition, identification of pathogenic mutations in MLL2 or KDM6A has a role in patients with intellectual disability or characteristic malformations.
KS is inherited in an autosomal dominant manner. The proportion of KS caused by de novo mutations is unknown, but is likely high based on clinical experience. Each child of an individual with KS has a 50% chance of inheriting the mutation. Prenatal diagnosis for pregnancies at increased risk is possible if the disease-causing mutation in an affected family member is known.
In summary, one novel mutation (c.5256_5257delGA;p.Lys1753 Alafs*34) was identified in a Korean patient with KS and it was revealed as a de novo mutaiton. The biological significance and pathogenicity of MLL2 in KS patients is not clearly demonstrated. Therefore, functionally-oriented studies are needed. Furthermore, other KS-associated genes need to be discovered.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


