


Introduction
Malformations of cortical development are rare congenital anomalies of the cerebral cortex that have long been of interest to neuroscientists and pediatricians. The term “malformations of cortical development” was first introduced in 19961,2). Patients with cortical malformations clinically present with epilepsy that is frequently intractable and various degrees of developmental delay and intellectual disability. It is assumed that approximately 40% of children with intractable epilepsy have malformations of cortical development3).
Malformations of cortical development encompass a broad spectrum of anomalous cortical formations with diverse anatomical and morphological abnormalities, a variety of genetic causes, and different clinical presentations. The emergence of brain magnetic resonance imaging (MRI) has greatly contributed to the determination of exact morphologies of cortical malformations. The hypothetical mechanisms of cortical malformation include any interruptions during the formation of the cerebral cortex: viral infections, genetic causes, vascular events, etcetera4). Recent developments in genetic analysis methods have improved our understanding of these pathological mechanisms, and offer implications for future therapeutic development. The present review will discuss the process of normal cortical development, the pathogenesis and classification of malformations, and the clinical diagnostic approach for malformations of cortical development.
Normal cortical development
The development of the cerebral cortex includes a series of complex processes. In the initial step, the proliferation (mitosis) of neuronal precursors in the ventricular zone results in an increase in the number of neurons. Next, neurons migrate from the ventricular zone to the cortex. The last step is the postmigrational organization of the neurons in the cortex5,6).
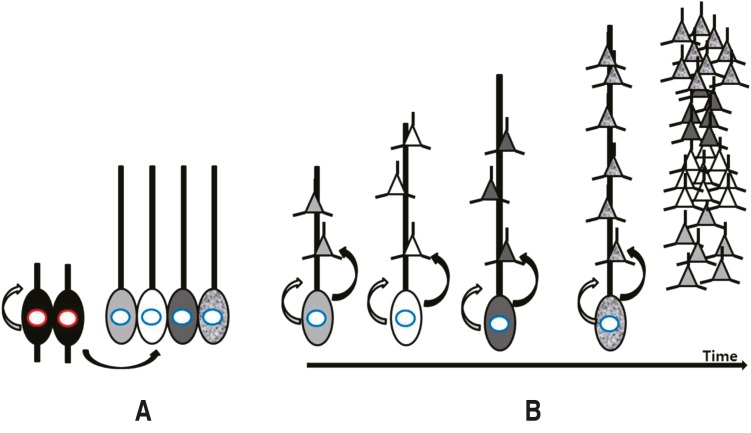
The mature cerebral cortex includes 2 types of cells: cortical neurons and glia. Neurons facilitate the signaling of the nervous system, and glia have supportive roles for the survival and function of neurons. There are 2 types of neurons: excitatory neurons and inhibitory interneurons. Excitatory neurons are born from polarized neuroepithelial cells (neuronal precursors) in the lateral ventricle. Excitatory neurons divide and differentiate into radial glial progenitors that extend fibers to the pial surface (Fig. 1)7). Radial glial progenitors can then divide into intermediate progenitors and outer radial glial progenitors, which divide and differentiate into excitatory projection neurons that migrate along the processes of radial glia to form the cortical plate. The cerebral cortex develops in an “inside-out” fashion. Early-born neurons form the deepest layers (V and VI) and later-born neurons form the more superficial layers of the cortex (II and III). Structural barriers of the pial surface and molecular signals have roles in the arrest of neuronal migration4). In this manner, the 6-layered laminar structure of the cerebral cortex is formed. Inhibitory interneurons are born and migrate tangentially to the dorsal telencephalon, turn and migrate radially to the cortical plate, and are organized with excitatory neurons8,9). Glia are formed from progenitor cells after neurogenesis9,10).
Pathogenesis and classification of malformations of cortical development
Malformations of cortical development result from the disruption of normal cortical development processes. In 1996, the initial classification scheme for cortical development was introduced based on the stage in which the developmental process was disturbed1). This scheme has since been updated and revised according to newly discovered mechanisms of pathogenesis; the most recent revision was published in 20122). There are 3 groups of cortical development malformation based on the timing and pathogenesis of the disruption: (1) group I, malformations secondary to abnormal neuronal and glial proliferation or apoptosis; (2) group II, malformations due to abnormal neuronal migration; and (3) group III, malformations associated with abnormal postmigrational development.
1. Group I: malformations secondary to abnormal neuronal and glial proliferation or apoptosis
Group I is associated with interruptions in the initial stages of neuronal precursor proliferation as well as disequilibrium between the proliferation and apoptosis of neuronal precursors. Subgroups include (1) group I.A: microcephaly; (2) group I.B: megalencephaly; and (3) group I.C: cortical dysgenesis with abnormal cell proliferation.
1) Microcephaly
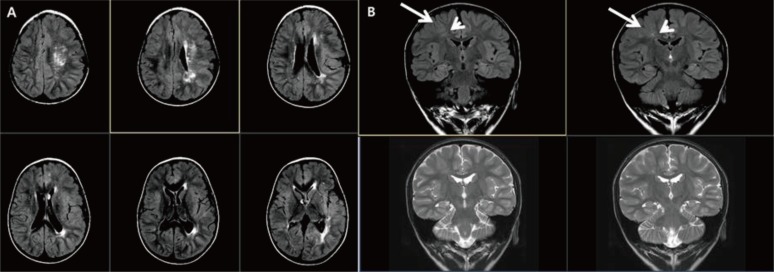
Microcephaly is defined by a head circumference less than 2 standard deviations below the average. Primary microcephaly, frequently referred to as microcephaly vera, is characterized by a small brain size without any anomalies in other organs. In primary microcephaly, brain MRI shows a normal structure, although the actual volume of the brain is less than 2 standard deviations below the average (Fig. 2A). Occasionally, a simplified gyral pattern, heterotopia, hypomyelination, or cerebellar hypoplasia can accompany microcephaly4). Microcephaly results from reduced proliferation or accelerated apoptosis. To date, most gene mutations known to cause microcephaly affect neurogenesis pathways by altering regulation of transcription, cell cycle, centrosome duplication and maturation, mitotic spindle disruption, and DNA repair defects (Table 1)2,11,12,13,14,15,16). These factors affect neurogenesis during phases of mitosis, which can produce proliferation abnormalities and result in a smaller population of neurons.
2) Megalencephaly
Megalencephaly refers to a condition in which the brain is more than 2 standard deviations above average (Fig. 2B). This group is not fully defined, and 6% of patients with polymicrogyria show megalencephaly17).
The discovery of specific pathways that control cell proliferation has informed the pathogenesis of several malformations of cortical development. The mammalian target of rapamycin (mTOR) pathway plays an important role in abnormal cortical development of the tuberous sclerosis complex (TSC)18). The discovery of the mTOR pathway's role in TSC has had great therapeutic implications for TSC: mTOR pathway inhibitors are now used in the treatment of subependymal giant cell astrocytoma19).
3) Hemimegalencephaly
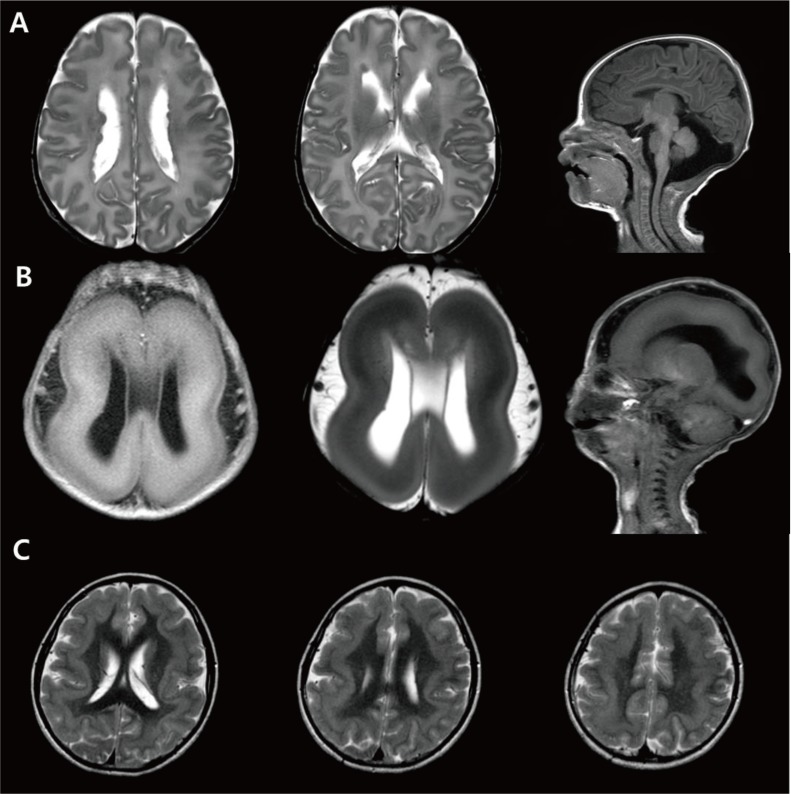
Hemimegalencephaly is the overgrowth of one brain hemisphere or part of a hemisphere. The pathology typically reveals various degrees of cortical dysplasia, white matter abnormalities, and polymicrogyria. Hemimegalencephaly can present as an isolated anomaly or it can exist as part of syndrome such as Klippel-Trenauney syndrome or hypomelanosis of Ito20). Patients with hemimegalencephaly present with mental retardation, early-onset intractable epilepsy, and contralateral hemiparesis. Brain MRI of these patients reveals abnormal enlargement of at least one lobe or one hemisphere, white matter signal abnormalities, pachygyria, and heterotopia (Fig. 3A)4). De novo somatic mutation of the AKT3 pathway was proposed to be associated with hemimegalencephaly in surgical specimens21,22).
4) Focal cortical dysplasia
Focal cortical dysplasia is one of the most frequent causes of intractable focal epilepsy. The pathology shows dysplastic neurons, balloon cells, and cortical dyslamination23,24). High resolution MRI can identify blurring of the gray-white matter junction, abnormal gyral thickening, and gray or white matter abnormalities (Fig. 3B)24,25). The pathogenesis of focal cortical dysplasia has yet to be revealed; however, brain somatic mutation of mTOR was recently proposed to underlie type II focal cortical dysplasia26).
2. Group II: malformations due to abnormal neuronal migration
Group II is divided into 4 subgroups according to the timing of neuronal migration interruption: (1) group II.A, periventricular heterotopia (problems in the initiation of migration); (2) group II.B, generalized abnormalities of transmantle migration (lissencephalies); (3) group II.C, localized abnormalities of transmantle migration (subcortical heterotopia); and (4) group II.D, anomalies associated with abnormal terminal migration or defects in pial limiting membranes (cobblestone malformations)2).
1) Heterotopia (group II.A)
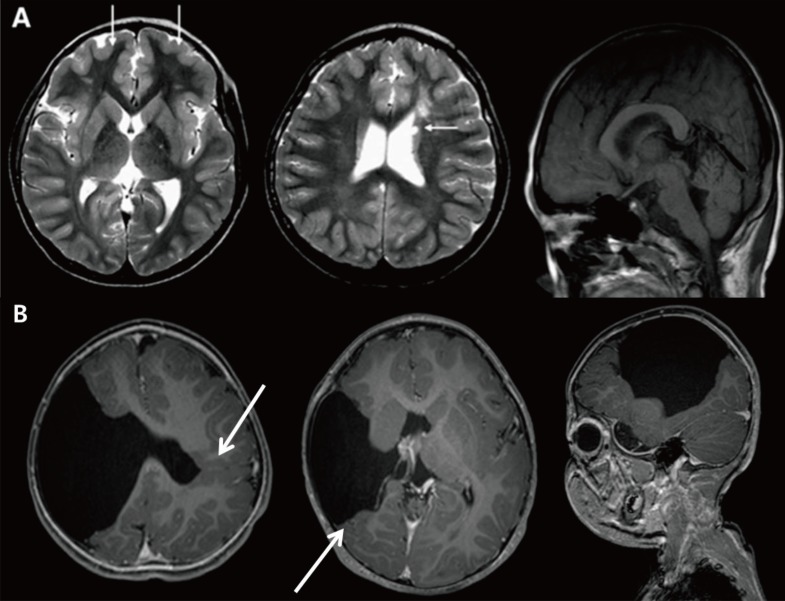
Periventricular heterotopia is characterized by neuronal nodules in the periventricular area and a normal cerebral cortex. Heterotopia results from the failure to initiate migration in a small group of neurons. Pathological specimens show normal-shaped neurons and glia accompanied by myelinated fibers and gliosis27). Approximately 90% of patients present with various types of seizures, mostly in adolescence. Brain MRI of these patients shows variously sized nodules along the lateral ventricles (Fig. 4A). The possible causes of periventricular heterotopia are genetic
2) Classical (type I) lissencephaly (group II.B)
Lissencephaly is one of the best-known malformations of cortical development, referred to as “smooth brain,” and results from the absence of normal gyri and sulci formation. Patients with type I lissencephaly have severe intellectual disability, microcephaly, and intractable epilepsy including infantile spasms. There are several genes that have been identified as causal to this malformation, including LIS1, DCX, TUBA1A, ARX, and RELN (Table 1)28,29,30,31). Brain MRI of these patients shows snowman-shaped configurations with areas of pachygyria and agyria (Fig. 4B). The posterior head region is more severely affected in patients with LIS1 mutations, while the frontal and temporal areas are smoother in patients with DCX mutations32).
3) Subcortical band heterotopia (group II.C)
Subcortical band heterotopia is also referred to as “double cortex” syndrome, because it is characterized by a band of subcortical heterotopic neurons between the ventricle and cerebral cortex. This disorder is mainly seen in females and is associated with intellectual disability and intractable epilepsy. Subcortical band heterotopia is known to be associated with mutations of the DCX gene33). The function of the DCX protein is to direct neuronal migration by regulating the organization and stability of microtubules that facilitate neuronal motility during cortico-genesis28). Mutation of the DCX gene in males causes classical lissencephaly. Brain MRI of these patients reveals a thin layer of white matter between 2 layers of gray matter (Fig. 4C).
4) Cobble stone malformations (type II lissencephaly, group II.D)
Type II lissencephaly is characterized by the nodular appearance of the cerebral cortex due to disorganization of the cortical layers, and excessive migration of neurons through the pial surface. This disorder is frequently associated with various eye abnormalities and congenital muscular dystrophies4). Clinically, increases in serum creatine kinase in patients with type II lissencephaly can expedite diagnosis. The clinical phenotype varies according to the causative genes. Fukuyama muscular dystrophy is the mildest form of type II lissencephaly, mainly existing in Japan and rarely in Korea (Fig. 5A)34,35). Muscle-eye-brain disease has been mainly reported in Finnish populations with early onset eye symptoms, intellectual disability, myoclonus, and congenital muscular dystrophy36). Walker–Warburg syndrome is the most severe form of type II lissencephaly and results in early infantile death37).
3. Group III: malformations secondary to abnormal postmigrational development
According to the classifications proposed in 2012, polymicrogyria is divided into 4 groups: group III.A, with schizencephalic clefts or calcifications that are presumably due to infection or vascular causes; group III.B, without clefts or calcifications, and may be genetic or disruptive; group III.C, as part of genetically defined multiple congenital anomaly syndromes; and group III.D, which is associated with inborn errors of metabolism2).
1) Polymicrogyria and schizencephaly (group III.A)
Polymicrogyria refers to a lumpy appearance of the cerebral cortex resultant from numerous small gyri separated by shallow sulci. This malformation can be focal or diffuse, and unilateral or bilateral. Bilateral polymicrogyria is frequently seen affecting the frontal, fronto-parietal, parieto-occipital, perisylvian, or medial occipital areas. Polymicrogyria is associated with various degrees of intellectual disability, motor dysfunction, and epilepsy. Familial polymicrogyria syndrome involves the fronto-parietal and perisylvian areas and is reported to be associated with mutation of GPR5638). Brain MRI of these patients shows small irregular gyri and vague gray/white matter differentiation.
Schizencephaly is characterized by the appearance of a cleft between the cortex and ventricle that is lined by polymicrogyric cortex (Fig. 5B). This malformation can be unilateral and bilateral; type I includes “closed-lip” clefts and type II includes “open-lip” clefts. The phenotype is more severe in type II, with patients showing developmental delay, microcephaly, intractable epilepsy, and motor deficits in the contralateral limbs4).
2) Focal cortical dysplasia (group III.C)
Focal cortical dysplasia is the most common cause of intractable focal epilepsy and is frequently considered a candidate for epilepsy surgery. Certain forms of focal cortical dysplasia are classified as group III because they can result from injury to the cerebral cortex in the later stages of cerebral cortical development. There have been reports about the various causes of mild forms of focal cortical development (type I), and these include prenatal and perinatal injuries such as severe prematurity, asphyxia, bleeding, hydrocephalus, and stroke39,40). In addition, group III.C cases are associated with physical injury, vascular malformation, or epileptogenic tumor by definition.
Diagnostic approach to malformations of cortical development
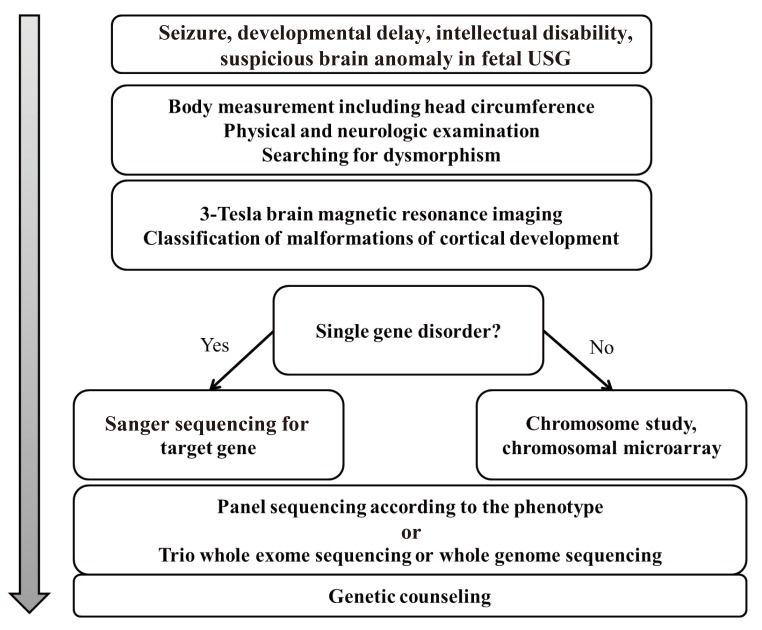
Patients with malformations of cortical development usually present with intractable seizures or developmental delay. The phenotype is extremely heterogeneous; the age of presentation, the degree of developmental delay, and the severity of neurologic deficit can vary according to the type of malformation. Clinicians diagnose malformations of cortical development following brain MRI as a diagnostic process for seizure or developmental delay. Then, a radiologic classification is made according to the morphological characteristics of the brain MRI. The diagnostic steps are summarized in Fig. 6. The dysmorphisms and anomalies accompanying cortical malformations require thorough investigation. If brain MRI findings or phenotype imply a certain genetic disorder such as periventricular nodular heterotopia, lissencephaly, or cobble stone malformations, Sanger sequencing for the candidate gene is the most direct method of diagnosis. If there are associated anomalies or dysmorphisms such as Miller-Dieker syndrome, a chromosome study or chromosomal microarray is necessary. If there are multiple candidate genes for a given cortical malformation group, such as microcephaly without other specific phenotype, targeted gene panel testing using next generation sequencing is more useful. If no causative gene is found by targeted gene sequencing, trio whole-exome sequencing for the proband and parents can be considered for a genetic diagnosis. Genetic diagnosis is recommended because it is a prerequisite for genetic counseling, explanation of the disease course, and prognosis. At present, Sanger sequencing for the causative genes of malformations of cortical dysplasia and targeted gene sequencing are not formally permitted by the Korean National Health Insurance Service. However, most genetic analyses are possible on a research basis, and these tests will be permitted formally in the near future.
Conclusion
Research investigating the genetic basis of various malformations of cortical development has improved our understandings of brain development and the pathophysiological basis of cortical malformations. Different mutations of the same gene can cause different phenotypic presentations depending on the degree of protein dysfunction. Loss-of-function mutations or gain-of-function mutations cause different phenotypes. Mosaicism can occur, revealing focal or milder phenotypic variations. In addition, functional mosaicism occurs due to X chromosome inactivation. In cases of gene mutation, causative management is not possible; however, elucidating causative genes and pathological mechanisms is a great step towards therapeutic development.
Malformations of cortical development have become increasingly recognized with the introduction of brain MRI. Cortical malformations are a major cause of intractable epilepsy and developmental delay. Genetic mutations and the resultant pathologies explain some malformations of cortical development, and these mechanisms are mainly associated with neuronal precursor proliferation, migration, and organization. Research about the pathogenesis of cortical malformation continues to elucidate the secrets of cerebral development. Diagnosis of malformations of cortical development will lead to better treatment for seizures and developmental delay, and contribute to genetic counseling for the family. Future studies of the pathogenesis of malformations of cortical development will enhance the development of treatments for various stages of brain development.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


