


Introduction
Early-onset epileptic encephalopathies (EOEE) are one of the most devastating early onset epilepsies that contribute to progressive decline of cerebral function1). Most patients show the three main features of EOEE: refractory seizures, severe electroencephalographic abnormalities, and developmental delay or intellectual disability2). A tendency to be refractory to antiepileptic drugs often leads to severe cognitive and behavioral impairment3). Identifiable primary causes, such as known structural, neurodegenerative, metabolic, genetic, or chromosomal disorders, and an increasing number of novel genetic causes are being identified in EOEE3,4). The most common causes are structural brain abnormalities and inborn metabolic defects5). If neuroimaging and biochemical examinations exclude common etiologies, the remaining fraction, comprising about one third of all EOEE cases, represents cryptogenic cases, where genetic factors are considered to have an important role6,7). EOEE is a genetically heterogeneous disorder: over 100 genes have been suggested to be involved in the etiology of these syndromes8). Many EOEE cases are sporadic, occurring in patients with no family history of seizures or epilepsies9). Sporadic cases are commonly caused by autosomal dominant de novo mutations in genes encoding neuronal proteins. EOEE can also be inherited in an autosomal recessive or X-linked manner. Next-generation sequencing (NGS) technology has revolutionized our ability to sequence DNA at the whole exome or whole genome level at an increasingly affordable price10). Whole exome sequencing now costs under $1,000 per sample and numerous studies have successfully identified de novo mutations in individuals with various neurodevelopmental disorders11,12). In epilepsy genetics, the focus has been almost exclusively on genes encoding membrane ion channel proteins. However, an increasing number of mutations in genes encoding proteins other than ion channels are now being identified by NGS2). Epi4k, the international consortium for advanced genetic studies of epilepsy, has been making significant progress in epilepsy genetics by using NGS. The consortium was launched in 2011 in response to a National Institute of Neurological Disorders and Stroke (NINDS) Funding Opportunity Announcement soliciting applications for "Centers Without Walls for Collaborative Research in the Epilepsies: Genetics and Genomics of Human Epilepsies" and adopted the name "Epi4K: Gene Discovery in 4000 Genomes"10,13). The first project of Epi4K focused on the discovery of de novo mutations in Lennox-Gastaut syndrome and infantile spasms. Over 300 de novo mutations in genes including SCN1A, STXBP1, SCN8A, SCN2A, CDKL5, GABRB3, ALG13, CACNA1A, CHD2, FLNA, GABRA1, GRIN1, GRIN2B, HNRNPU, IQSEC2, MTOR, and NEDD4L have been discovered. Statistical evidence of association between epileptic encephalopathy and mutations in GABRB3 and ALG13 was also identified9,10). In this article, we will focus on the diagnostic strategies for EOEE, especially on NGS-based genetic analysis for cryptogenic EOEE cases.
Epileptic encephalopathy syndromes in infancy
1. Vitamin-responsive epileptic encephalopathies
Vitamin-responsive epileptic encephalopathies are rare but important causes of EOEE. They commonly result in refractory seizures with poor neurocognitive outcomes if specific treatment is delayed. The type of seizures, etiologies, and treatments are summarized in Table 1. If seizures persist despite the use of two or more appropriate anticonvulsants at maximum tolerated doses, then add-on vitamin treatment should be considered. Pyridoxine-dependent epilepsy (PDE) and pyridoxal-5-phosphate-dependent epilepsy share similar clinical presentations as pyridoxal-5-phosphate is derived from pyridoxine14). In the early onset type, patients will typically develop abnormal movements soon after birth and electroencephalogram (EEG) can be either completely normal or exhibit a burst suppression pattern15). Prognosis is generally good with prompt treatment, but death or significant intellectual and motor disability may occur if treatment is delayed16). Pyridoxal-5-phosphate may be used as an initial treatment, which would be effective against both PDE and pyridoxal-5-phosphate dependent epilepsy. Pyridoxine could be introduced later, replacing pyridoxal-5-phosphate as a cheaper and similarly effective alternative, if PDE diagnosis is confirmed17). Folinic acid responsive epilepsies are caused by low concentration of 5-methyltetrahydrofolate (MTHF) in the cerebrospinal fluid (CSF), which is associated with various neurological conditions18). Genetic or autoimmune mechanisms cause cerebral folate deficiency and delayed treatment may lead to encephalopathy with severe learning disabilities. EEG may show abnormal background activity with multifocal spike-wave complexes, but typically has no diagnostic features. Neuroimaging results are also usually normal19). Patients either do not respond to pyridoxine at all or exhibit only a temporary improvement. However, such patients show a marked neurological recovery including cessation of seizures upon folinic acid treatment18). Biotinidase deficiency is a biotin-responsive metabolic disorder causing impairment of functions of multiple carboxylases and presenting with seizures, hypotonia, visual/auditory symptoms, eczema, and alopecia. Untreated children usually have neurocutaneous features between the ages of 2 and 5 months18,20). Seventy percent of patients have various type of seizures including infantile spasms21). Seizures and other symptoms improve often within a day of treatment with biotin. It has been suggested that similarly to pyridoxine trials, biotin treatment should be considered in any child with poorly controlled seizures22). Regardless of age or weight, a dose of 5- to 20-mg biotin daily has been found to be effective and needs to be continued for the rest of patient's life if it offers stable improvement23,24). Vitamin B12 deficiency may lead to various neurologic symptoms including developmental delay or regression, irritability, weakness, hypotonia, and convulsions. Seizures are a rare presentation of vitamin B12 deficiency but they have been occasionally reported, especially in infants, including cases of West syndrome25,26). Vitamin B12 deficiency caused by dietary preferences of mothers, who may be vegan, is the most common cause of such symptoms in breast-fed infants between 4 and 8 months, even when the mothers exhibit no hematological or neurological symptoms themselves26,27). Serum levels of methylmalonic acid and total homocysteine have been shown to be markedly elevated in the majority of such patients. Vitamin B12 deficiency should be considered in all infants with developmental delay, hypotonia, or seizures for whom an alternate diagnosis cannot be made.
2. Ohtahara syndrome
Ohtahara syndrome is often defined as an early infantile epileptic encephalopathy with a characteristic EEG pattern, suppression-burst, during which higher-voltage bursts of slow waves mixed with multifocal spikes alternate with isoelectric suppression phase28). EEG shows a continuous suppression-burst pattern in both waking and sleeping states. The onset is between neonatal period and early infancy, usually within the first 3 months of age, with some mothers reporting seizure-like movements of the fetus during pregnancy29). Etiologically, structural brain lesions, such as diffuse subependymal band heterotopia or midbrain dysplasia, are the most probable causes of Ohtahara syndrome30,31). Predominant seizures detected in Ohtahara syndrome patients are repetitive, frequent, tonic spasms occurring with or without series formation, although other seizure types can also be observed29,32). Antiepileptic and immunomodulating drugs are generally ineffective in treating tonic spasms, although rare cases show improvement upon such treatment. In general, prognosis is extremely poor with chronic intractable seizures and severe psychomotor retardation. Seizure patterns usually change with time: frequently cases evolve to West syndrome and further to Lennox-Gastaut syndrome with age. Nearly 50% of affected children are likely to die in infancy or childhood32,33).
3. Early myoclonic epileptic encephalopathy
Early myoclonic encephalopathy (EME) is characterized by fragmentary myoclonic jerks or violent myoclonic spasms, which generally occur in the neonatal period or early infancy. Partial myoclonus and partial motor seizures are the main seizure types in EME, but generalized myoclonus can also be observed in some patients. Partial motor seizures are frequent. They occur shortly after erratic myoclonus and shift typically from one part of the body to another in a random, asynchronous pattern34). The myoclonus usually involves the face or extremities, but may be restricted to some other part of the body. Typical interictal EEG shows a suppression-burst pattern similar to that seen in Ohtahara syndrome35). Suppression-bursts become more apparent in sleep and may persist until late childhood after a transient evolution to hypsarrhythmia in the middle to late infancy31). The generalized myoclonic jerk typically is associated with a generalized or fragmentary burst of polyspike, spike, and slow wave discharges, but erratic myoclonia may or may not be related to the bursts35). The etiology is variable and often remains unknown, but nonstructural/metabolic disorders are most probable causes of EME. Vitamin responsive epilepsies, such as PDE, pyridoxal-5-phosphate-dependent epilepsy, or folic acid responsive epilepsy can show typical clinical and EEG features of EME. Other inborn metabolic deficiencies, such as nonketotic hyperglycinemia, methylmalonic acidemia, or propionic acidemia, can also demonstrate EME features. Concentrations of serum amino acids and urine organic acids, as well as amino acid content of the CSF should be analyzed in patients with EME34,36). The prognosis for EME is also poor and there is no effective treatment except for vitamin responsive epilepsies. EME persists for long periods without evolution, except for the occasional transient phase of West syndrome, or changes into partial or severe epilepsy with multiple independent spike foci31).
4. Infantile spasms (West syndrome)
Infantile spasms or West syndrome is the most common epilepsy syndrome in infancy, which presents with a combination of the triad of infantile spasms, developmental deterioration, and hypsarrhythmic EEG pattern3,37). Typically, the spasms involve brief symmetrical contractions of musculature of the neck, trunk, and extremities, which frequently occur in clusters38,39). An individual spasm lasts for seconds (usual duration 1-2 seconds) and is often longer than typical myoclonus (duration up to 200 ms), though not as long as tonic seizures, which last for several seconds. Spasms are usually recurrent with a period of 5-30 seconds3,34). The spasms may be subtle and isolated at onset, typically clustering with time. Patients typically exhibit several clusters per day, particularly during drowsiness37,40). Hypsarrhythmia, a typical interictal EEG pattern observed in this condition, consists of a disorganized pattern with asynchronous, very high amplitude multifocal spike and sharp wave discharges. The etiology can be classified into symptomatic and cryptogenic cases. The fraction of symptomatic cases has been steadily increasing due to improved diagnostic techniques, such as metabolic and genetic testing, as well as neuroimaging41). Symptomatic causes are found in nearly 60% of cases, which include cerebral malformations, infection, hemorrhage, hypoxic-ischemic injury, metabolic disorders, and genetic conditions40,42). Tuberous sclerosis complex (TSC) is an important cause of infantile spasms and 75%-80% of individuals with TSC may develop epilepsy41). Outcomes are mostly dependent on the etiology; cryptogenic patients who are treated early have more favorable prognosis than patients with symptomatic varieties43,44).
5. Malignant migrating partial epilepsy in infancy
Malignant migrating partial epilepsy in infancy (MMPEI) is characterized by neonatal or early infantile onset migrating partial seizures, which usually last a few weeks or months, and the frequency becomes nearly continuous with time. Patients show frequent partial seizures of multifocal onset with autonomic manifestations, such as apnea, flushing, or cyanosis45). The interictal EEG shows multifocal epileptiform discharges with diffuse slowing of background activity. The multifocal discharges poorly activated by sleep in all cases and background activity slows down with fluctuating asymmetry between different recordings34). Most cases have no clear etiology of structural or biochemical abnormalities suggesting contribution of genetic factors. However, genetic tests usually fail to detect mutations in KCNQ2, KCNQ3, SCN1A, SCN2A, or CLCN2 genes in MMPEI46). Seizures are often intractable and global developmental delay is common. Most patients develop an acquired microcephaly by the end of the first year of age and a number of patients die before that time or later, in the course of the follow-up period45).
6. Myoclonic status in nonprogressive encephalopathies
Myoclonic status in nonprogressive encephalopathies (MSNE) is an early onset epileptic syndrome characterized by dulling of consciousness and responsiveness with or without jerks, which may last for hours, days, or weeks47). Interictal EEG consists of multifocal epileptiform discharges and background slowing. Ictal EEG recording may demonstrate generalized slow spike and wave or an absence pattern, depending on the seizure type34). A genetic cause, such as Angelman syndrome or 4p syndrome, is found in approximately half of the children. Other reported structural causes include hypoxic-ischemic injury and cortical dysplasia34). Most children are resistant to different therapies, even to intravenous benzodiazepines, and the status may become a life-threatening event47). Seizures often persist into adulthood and the final outcome is very poor with developmental regression and severe mental retardation, especially in patients with repeated episodes of myoclonic status48,49).
7. Dravet syndrome (severe myoclonic epilepsy in infancy)
Dravet syndrome is a genetically determined severe epileptic encephalopathy, which begins in the first year of life in an otherwise normal infant. The epilepsy starts with seizures, which may not initially differ from those associated with febrile illnesses. Even mild fever is an important trigger factor, but some cases are provoked by a nonfebrile illness, immunization, or hot environment50). It is not easy to differentiate these children from others with febrile convulsions, who will eventually get better and will not develop other types of seizures. During the second year of the life, seizures become more frequent, persistent, and often more lateralized. At that stage, seizures no longer occur only when a child has high temperature, but can happen at any time of the day34). Seizures are complex febrile, afebrile generalized, unilateral clonic, or tonic-clonic. The condition evolves to other types, such as myoclonic, atypical absence, complex partial seizures, and frequent status epilepticus50). EEG is usually normal at early stages of this condition. However, by the time a child is 2 years old, epileptic discharges with spike and wave or polyspikes are observed, which occur either as single events or in bursts. Prognosis is very poor, as this syndrome is associated with developmental delay, cognitive dysfunction, and behavioral problems.
Diagnostic approaches to underlying causes of EOEE
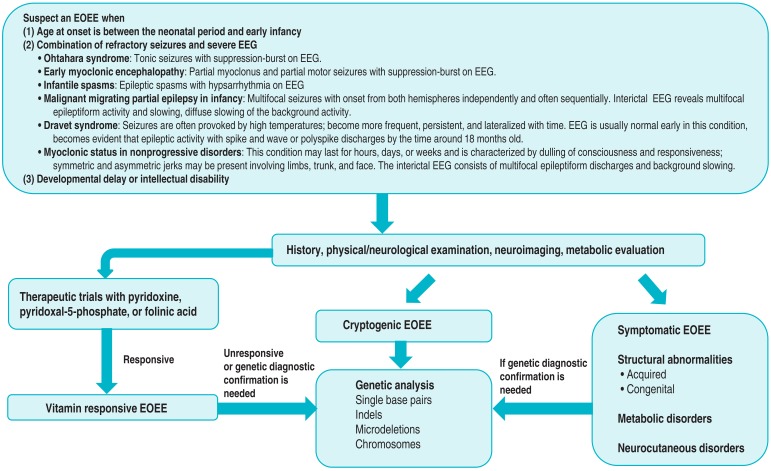
Diagnostic approaches to underlying causes of EOEE are summarized in Fig. 1. Assessments begin with the elucidation of the history and semiology of clinical seizures and analysis of EEG findings. When age onset, clinical manifestations, and EEG findings are consistent with EOEE, an evaluation of possible underlying etiologies should be performed. EOEE must be distinguished from acute symptomatic seizures occurring in infancy, for example, those caused by infection, hypoglycemia, or electrolyte disturbance3). If the primary investigations exclude precipitating conditions, a trial with the administration of a vitaminic compound (pyridoxine, pyridoxal-5-phosphate, or folinic acid) should then be initiated51,52). These new insights re-emphasize the importance of early treatment of neonatal seizures with vitamins, whatever the suspected cause. Pyridoxine, folic acid, and cyanocobalamin are still the most commonly prescribed treatments, largely due to their commercial availability and affordability. However, safer and more effective formulations can be obtained. Ideally, pyridoxal phosphate can be used instead of pyridoxine, but it is not licensed for sale or is not easily available in many countries. Direct purchasing biologically active vitamins overseas is possible through websites via internet searching. When importing 7 or more bottles of vitamins, import declaration and a medical prescription is necessary because vitamins are categorized as health functional foods in Korea. In the case of purchasing less than 7 bottles of Korean Ministry of Food and Drug Safety unrestrained vitamins for personal use, however, it is possible to pass customs without any prescriptions or declaration. Older children presenting with recurrent febrile status epilepticus or intractable seizures should receive similar therapy and, in addition, biotin in order to exclude biotinidase deficiency. If the patient responds to treatment, suitable biochemical and genetic investigations should then be undertaken to define the cause53). Clinicians should be aware that a poor response does not completely exclude diagnosis of vitamin dependent epilepsies51,54).
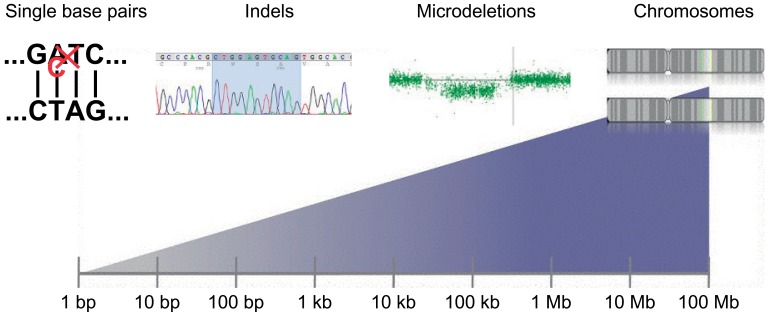
During physical/neurologic examinations, certain clinical parameters, such as the combination of dysmorphic features, neurologic deficits, or cutaneous lesions, may at that point suggest underlying etiology. Following the completion of anamnesis, physical/neurologic examinations, EEG analysis, and magnetic resonance imaging of the brain, approximately two-thirds of patients will have an established etiologic diagnosis without the need to conduct extensive metabolic testing41). The remaining patients with undetermined etiologies should be considered for further evaluation, which will depend on individual circumstances. Typical tests may include organic acids in urine, amino acids in serum, determination of biotinidase, analyses of neurotransmitters, lactic acid, amino acids, folate metabolites, and glucose in the CSF as well as other biochemical tests suggested by the patient's clinical course and study findings41). Although they are relatively rare, metabolic encephalopathies are important to be recognized. Patients with some of these syndromes can respond to specific treatments, but some antiepileptic drugs interfering with metabolic pathways may worsen the clinical condition, so specific genetic counseling should be provided in such cases55). In a recent study, diagnostic genetic testing for childhood epileptic encephalopathy found genetic causes in 28% of the patients: 7% had inherited genetic metabolic disorders, while 21% had other genetic causes including genetic syndromes, pathogenic copy number variants (CNVs) revealed by comparative genomic hybridization arrays, and epileptic encephalopathy related to mutations in the SCN1A, SCN2A, SCN8A, KCNQ2, STXBP1, PCDH19, and SLC9A6 genes56). Genes mutated in early onset epileptic encephalopathy are summarized in Table 2. Targeted next-generation sequencing panels increased genetic diagnostic yield from less than 10% to over 25% in patients with epileptic encephalopathy56). Identification of mutations underlying cryptogenic EOEE needs to be performed in a targeted fashion by sequencing the most likely candidate genes. Massive parallel sequencing approaches enabled obtaining sequence information directly, however, the abundance of novel data, be it on the level of common single nucleotide polymorphisms, CNVs, or overall sequence, has led to the identification of a vast amount of benign variation in the human genome (Fig. 2)57). Massive parallel sequencing in epilepsy genetics can be divided into three different fields: family studies, gene panel studies, and patient-parents trio studies. Family studies are performed to identify the causal monogenic variant in families. Panel studies trade additional genetic information for deeper coverage outside the selected genes in contrast to exome sequencing studies. Patient-parent trio studies focus on the genome-wide identification of de novo mutations57).
Conclusions
In conclusion, the diagnostic procedure for EOEE is still challenging, but early recognition and proper management have an important effect on its long-term outcomes. We believe that next-generation sequencing technologies are inspiring hope in a large number of children with cryptogenic encephalopathies and the use of these approaches will eventually lead to the development of new therapeutic strategies and, as a result, to more favorable long-term outcomes.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


