

Introduction
Chronic myelogenous leukemia (CML) is a very rare disease in children, with less than 10% of all CML cases diagnosed in children and adolescents. CML accounts for 3-5% of all childhood leukemias, and has an annual incidence of 1 in 1,000,0001).
CML is a myeloproliferative disorder defined by the presence of the Philadelphia chromosome, which arises from the reciprocal translocation of genes on chromosomes 9 and 22. It is characterized by the proliferation of a malignant clone containing the Philadelphia chromosome, resulting in bone marrow hyperplasia and peripheral blood leukocytosis and thrombocytosis. The Philadelphia chromosome in CML, t(9;22)(q34;q11), was named in 1960 after the city in which it was first discovered. It consists of a juxtaposition of breakpoint cluster region (BCR) gene on chromosome 22 with Abelson leukemia virus (ABL) gene, resulting in a fused BCR-ABL protein with constitutive tyrosine kinase activity, and subsequent activation of cytoplasmic and nuclear signal transduction pathways including STAT, RAS, JUN, MYC, and phosphatidylinositol-3 kinase. The ultimate result of such activation is the myeloid proliferation and differentiation and suppressed apoptosis that is characteristic of CML4).
CML is clinically divided into chronic phase (CP), accelerated phase (AP), and blast phase (BP). Most cases of CML are diagnosed in the CP, but about 10% are diagnosed in the advanced phases. Forty to fifty percent of the patients diagnosed in the CP are asymptomatic. Common symptoms include asthenia, splenic discomfort, weight loss, and bleeding5). With progression to advanced phases, other symptoms such as fever, night sweats, abdominal distension, and bone pain may become more evident, together with complications of leukostasis such as neurologic abnormalities, papilledema, retinal hemorrhage, tachypnea, and priapism.
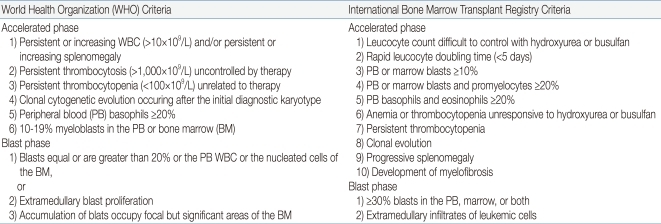
Similar to adults, children progress from the CP to AP, and finally to the BP, but 25% of the patients move directly from the CP to BP. Although the clinical and morphological boundaries of each phase may be blurred, the recognition of disease progression from the CP to either AP or BP is important for both the treatment and overall prognosis of the patient. Table 1 shows the WHO criteria for the classification of AP and BP6). The phenotype of BP is 60-80% myeloid and 20-30% lymphoid, with a small proportion showing mixed phenotype.
Diagnosis is made by complete blood count and bone marrow examination, with the identification of the characteristic Philadelphia chromosome by metaphase studies, fluorescent in-situ hybridization (FISH), and reverse-transcriptase polymerase chain reaction (RT-PCR). The bone marrow tends to be hypercellular. Approximately 50-70% of the patients have a WBC count above 100,000/mm3 in the peripheral blood, 30-50% have thrombocytosis, and 20% have anemia. The peripheral blood smear shows a broad range of myeloid cells that result from myeloid differentiation, including an increase in basophils and eosinophils. Serologic examination is notable for an increase in uric acid, lactate dehydrogenase, vitamin B12, and vitamin B12-binding protein (transcobalamin 1).
Allogeneic hematopoietic stem cell transplantation (HSCT) was previously considered the only curative treatment available. However, perspectives on treatment changed dramatically in 1996 with the development of imatinib mesylate (STI 571, Gleevec), a tyrosine kinase inhibitor (TKI). Currently, treatment with TKIs, including imatinib, has replaced HSCT as the primary treatment in the adult CML population, and its role is becoming more prominent in childhood CML7-9). In this review, we discuss the optimal strategies to cure childhood CML in the era of imatinib treatment.
Non-transplant therapies
1. The pre-imatinib era
In the 1950s and 1960s, the only medications available were busulfan and hydroxyurea, and the aim was cytoreduction and a decrease in hepatosplenomegaly rather than a durable cure of CML10). In the mid 1980s, the introduction of interferon-α (IFN-α) as a treatment for CML-CP resulted in 70-80% hematologic remission and 20-30% complete cytogenetic response. The efficacy of this drug led to numerous attempts to lengthen the response rate and duration through combination treatment with IFN-α and hydroxyurea, or IFN-α and low-dose cytarabine11-13). Reports of IFN-α treatment for childhood CML are limited, but Millot et al14) undertook a study of 14 patients, of whom 7 (50%) showed a complete hematologic response and 7 showed a major cytogenetic response, including 2 patients who showed a complete cytogenetic response (CCyR). However, a comparative study of TKI versus IFN-α among adults established the superiority of TKI and its firm role in the primary treatment of CML15).
2. The imatinib era
The treatment of CML has changed dramatically over the past few years. In the past, HSCT was the only known curative therapy and was the recommended treatment. In recent years, complete responses have been documented with TKIs directed against BCR-ABL, and front-line use of a BCR-ABL targeting TKI is now the standard treatment for most patients with CML. Imatinib, the first TKI to be developed, is a small-molecule inhibitor of various tyrosine kinases, including ABL tyrosine kinase, c-KIT, and plateletderived growth factor. In 1996, Druker et al16) initially reported on the specific cytotoxic effect of imatinib on proliferating myeloid cell lines specifically harboring BCR-ABL. Subsequent phase I and II trials verified the clinical efficacy of the drug, resulting in approval by the US Food and Drug Administration (US FDA) in 2001. The results of a phase III trial, which enrolled 1,106 patients, underscored the role of imatinib as the initial non-transplant treatment choice for newly diagnosed CML-CP patients; 87% of the patients treated with imatinib showed CCyR at 18 months with 3.3% disease progression, compared to 14.5% CCyR and 8.5% disease progression in patients treated with IFN-α and cytarabine. In addition, imatinib was better tolerated than the combination therapy17).
The results of clinical trials with imatinib in the adult patient population have been applied to children, and imatinib is now also the front-line treatment for childhood CML. Since the approval by the US FDA in 2003, several reports have been published on the effects and toxicities of imatinib in children. With regard to the dosage of imatinib, pediatric doses of 260 mg/m2 and 340 mg/m2 have been found to be on par with 400 mg and 600 mg, respectively; therefore, the recommended starting dose in children is 300 mg/m2 once daily (maximum absolute dose, 400 mg) and 400-500 mg/m2 for advanced-stage disease18, 19).
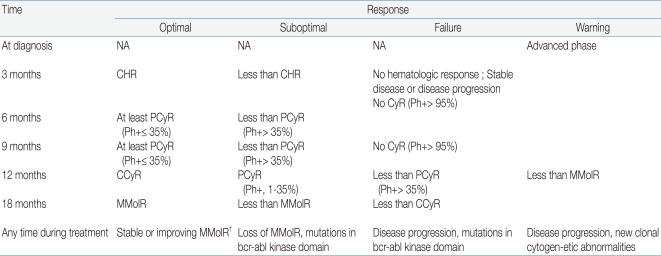
The evaluation of the response to TKI treatment is made through hematologic, cytogenetic, and molecular testing (Table 2)9, 20, 21). Recently, the European LeukemiaNet has published specific criteria on when and how to make response evaluations9). Hematologic response is assessed by complete blood count, whereas differential cytogenetic response (CyR) is assessed by analyzing at least 20 marrow metaphase cells and molecular response (MolR) is measured by checking BCR-ABL transcript levels using RT-PCR. The overall evaluation should lead to a classification of treatment response as optimal, suboptimal, or failure (Table 3)9, 22). The lack of any specific guidelines for children prompts the consideration of such adult-based criteria to measure response and decide upon clinical status such as imatinib resistance, intolerance, noncompliance, or disease progression. In patients with optimal response to imatinib, the drug may be continued until allogeneic HSCT is undertaken. In those who fail to respond, second-generation TKIs and HSCT need to be considered. In suboptimal responders, imatinib may be continued, possibly at a higher dosage, or second-generation TKIs may be introduced. Currently, clinical trials are under way in order to expedite the introduction of second-generation TKIs to the front-line treatment of childhood CML23).
Imatinib treatment may result in diverse side effects, but most are of mild to moderate severity, and possibly lesser so for children24-26). Common toxicities include nausea, vomiting, diarrhea, skin rash, edema, elevated liver enzymes, and cytopenia. A common complaint among children is nausea, and food and liquid intake is recommended with drug ingestion in order to minimize gastrointestinal tract toxicity, as food absorption has little effect on drug absorption. Lethargy, weight gain, myalgia, and cramps may also be significant side effects, and bone pain may require analgesics and anti-inflammatory agents. Of special consideration in children is the detrimental effect of imatinib on height growth and bone metabolism, which require long-term assessment of physical parameters as well as serum calcium, phosphorus, parathyroid hormone, and bone metabolic markers. However, little has thus far been reported on the incidence and treatment of complications resulting from prolonged imatinib use in children27-31).
Cases precluding treatment with imatinib include drug resistance and intolerance due to hematologic or non-hematologic toxicities. Resistance can be further divided into primary and secondary (relapse) types, which are characterized by lack of response from the start and loss of initial response to treatment, respectively. Mechanisms of resistance are varied and include mutations in the BCR-ABL kinase domain that prevent imatinib binding, BCR-ABL amplification or overexpression, clonal evolution of disease, and decreased drug bioavailability32).
3. Transplantation
Allogeneic HSCT was the only curative option in the pre-imatinib era. However, the good outcomes of imatinib treatment and accurate monitoring of minimal residual disease (MRD) has relegated HSCT to salvage therapy for patients with early signs of disease progression or resistance. For patients without a human leukocyte antigen (HLA)-matched sibling donor (MSD), the search for an unrelated donor should begin at diagnosis, since several months may pass before an appropriate donor is identified. The optimum time for HSCT is during the CP before progression to the AP or BP33-35). Outcomes of HSCT in the BP are poor, with less than 20% long-term survival. Therefore, it is important to undertake treatment with chemotherapy or TKIs to achieve at least hematologic response and to schedule HSCT as soon as possible, with a less than fully matched donor if necessary, in order to improve outcomes. Treatment of the BP that initially may have led to complete molecular response with TKIs can rapidly progress to overt relapse; this emphasizes the need for more frequent monitoring of disease progression in these patients than in those treated at the CP.
Limited comparative studies are available concerning the best conditioning regimen for childhood CML patients36). In theory, reduced intensity conditioning (RIC) should limit toxicity and preserve fertility in these patients. However, patients who received RIC during the advanced phase of disease may show a high rate of relapse, and such patients should be monitored on a short-term basis to predict the need for donor lymphocyte infusions (DLI) or reinitiation of TKI treatment. If allogeneic HSCT were to be reserved for patients who have failed imatinib, then relapse after RIC may prove difficult to manage with TKIs because of resistance. Treatment with early DLI, however, may lead to significant graft-versus-host disease (GvHD) in these patients. Hence, the efficacy of RIC in childhood CML has yet to be proven.
The most difficult scenario is in the case of children who have not responded to imatinib and also lack appropriate donors. Treatment without transplantation would require administration of drugs used in the past such as IFN-α, hydroxyurea, or low-dose cytarabine, but life prolongation would be difficult. These medications, however, may be used to lengthen survival until other experimental, targeted therapeutic measures that are currently being tried on adult patients become available for children. Other options include alternative transplantation measures such as cord blood transplantation and haploidentical transplantation.
Risk of relapse after HSCT in CP is relatively high at 20%, emphasizing the need for MRD monitoring. The effects of TKI treatment for the prevention of relapse after HSCT are still unclear, and require future prospective studies. Methods of treatment after relapse are without consensus, but further remissions may be obtained with DLI and TKI treatment. Imatinib treatment failure before HSCT may require dasatinib for relapse after transplantation.
There is general agreement that for adults, HSCT should be considered after trials of second-line treatment. For children, the most appropriate time for HSCT remains controversial. Whether HSCT should be considered as a first-line treatment when an MSD is not available and what should be the duration of imatinib treatment before HSCT are questions that remain to be answered. Treatment selection may also depend on national resources as well treatment efficacy. Developing countries with limited financial resources may favor one major curative attempt such as HSCT rather than lifelong treatment with an expensive drug. Advanced-phase patients would need to show hematologic or cytogenetic response before undergoing HSCT as first-line treatment37). The question of how long imatinib should be given to CML patients is, as yet, unanswered. Recently, Mahon et al38) showed that 38% of patients maintained complete molecular response after imatinib was stopped for 24 months. Further, patients with molecular relapse responded well to reinitiation of imatinib treatment. The data are promising, because they show the possibility of either intermittent imatinib dosing or complete withdrawal of imatinib, but extrapolation of these findings to children is premature.
Conclusion
Treatment of CML is a model of targeted therapy based on disease pathogenesis and effective monitoring of MRD to improve patient survival. In adults, allogeneic HSCT is no longer considered for patients in the CP, and in children, its role is gradually being replaced by TKI treatment. However, the objective for treatment of childhood CML is not palliation, but cure. Hence, the possible adverse effects that stem from long-term TKI treatment and the potential for disease progression while on TKIs weigh more heavily in the childhood CML population. The development of novel drugs with greater efficacy and less toxicity may allow these patients to be free of the burden of HSCT, with its inherent risk of transplant-related mortality and significant long-term complications. The introduction of new medications into the treatment regimens of children and international studies concerning the efficacy of these medications should take place as soon as possible.

 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


