

Introduction
Mitochondria produce energy to maintain cellular functions through the oxidative phosphorylation of mitochondrial respiratory chain complex (MRC) that exists within its inner membrane1, 2). Mitochondrial dysfunctions can happen in virtually any type of tissue or any organ, though they most commonly present with abnormal symptoms related with the neuromuscular system3, 4). In short, they present with a variety of symptoms depending on the organ they have involved5, 6).
Thus far, there have been many reports on the ophthalmologic involvement of mitochondrial diseases (MD)7, 8). A variety of ocular symptoms and signs, such as pigmentary retinal degeneration, external ophthalmoplegia, and optic atrophy, are observed in patients with MD. Optic nerve, retina, pigment epithelium, and ocular tissues are reported to be dependent on oxidative energy production9, 10), and some relationship between specific ophthalmologic diseases and MD have also been suggested11-13).
While there have been many reports and experiments published on ophthalmologic manifestations of MD, few studies have been conducted on the related factors themselves that affect the manifestation. This research tried to analyze the ophthalmologic symptoms and ophthalmologic examination (OE) results of patients with MD to better understand their relationship.
Materials and methods
1. Patients
The study was conducted with seventy-four patients who had been diagnosed as having MRC defect at the Gangnam Severance Hospital from September 2005 to March 2008. The study was approved by the institutional review board of our hospital and written informed consent was obtained from all patients. Evaluation of MRC dysfunction was performed by analyzing the activities of reduced nicotinamide adenine dinucleotide (NADH)-coenzyme Q (CoQ) reductase (complex I), succinate-CoQ reductase (complex II), succinate-cytochrome c reductase (complex II-III), cytochrome c reductase (complex III), cytochrome c oxidase (complex IV), oligomycin-sensitive ATPase (complex V), and citrate synthase assessed from isolated mitochondria in muscle tissue through use of standard spectrophotometric assays as described by Rustin, et al14). We defined MRC defect as reduction of residual enzyme activity below 20% of controls.
2. Clinical evaluation
Ophthalmologic symptoms were initially evaluated by inquiring about the subjects' medical history, followed by a neurological examination. Ophthalmologists performed an OE with a funduscopy after dilating the pupils. The seventy-four patients were divided into two groups based on the results of fundus examination as either a normal or abnormal group. The two groups were then compared and analyzed based on their clinical features, biochemical test, morphological analysis, and neuroimaging findings.
Lactic acidosis was diagnosed as a serum lactate higher than 2.2 mmol/L. Increased lactate/pyruvate ratio was defined when it was higher than 20, and cerebrospinal fluid (CSF) lactate levels when it was higher than 22 mg/dL. Urine organic acid metabolites were interpreted as a mitochondrial dysfunction when lactic aciduria was accompanied with an elevation of Kreb cycle metabolites, such as 3-methylglutaconic acid, ethylmalonic acid, dicarboxylic acid, tiglylglycine, butyrylglycine, 2-ethylhydracrylic, 2-methylsuccinate, and isovalerylglycine. In plasma amino acid assays, special attention was paid to the secondary hyperalaninemia derived from lactic acidemia. The mitochondrial fatty acid disorder was analyzed through the evaluation of plasma acylcarnitine profile screen15, 16).
In the morphological analysis, light microscopic observation confirmed ragged red fibers by a modified Gomori trichrome stain. Succinate dehydrogenase, NADH, ATPase, Periodic acid Schiff (PAS), and lipid stain were all performed for the immunohistochemical examination. Electron microscopic observation confirmed abnormal mitochondrial structures, including the proliferation of the cristae mitochondriales, paracrystalline inclusions, or mitochondrial proliferation with an accumulation of excessive or enlarged mitochondria in the subsarcolemmal regions17).
Results
1. Ophthalmologic symptoms & examination
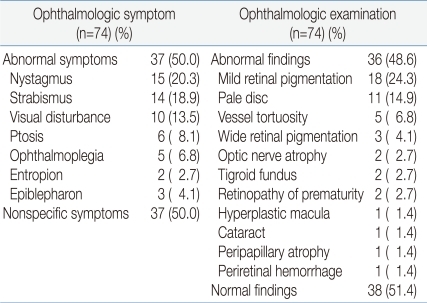
Thirty-seven (50%) out of the seventy-four MD patients developed ophthalmologic symptoms. Abnormal findings were observed in thirty-six (48.6%) patients during the OE, and sixteen (21.6%) of them had not experienced ocular symptoms. Among patients with ophthalmologic symptoms, fifteen patients (20.3%) had nystagmus, fourteen had (18.9%) strabismus, ten had (13.5%) visual disturbance, six had (8.1%) ptosis, and five had (6.8%) opthalmoplegia. Among the patients with abnormal findings in the OE, eighteen patients (24.3%) had mild retinal pigmentation, eleven had (14.9%) pale disc, five had (6.8%) vessel tortuosity, three had (4.1%) wide retinal pigmentation, two had (2.7%) optic nerve atrophy, two had (2.7%) tigroid fundus, two had (2.7%) retinopathy of prematurity, one had (1.4%) hyperplastic macula, one had (1.4%) cataract, peripapillary atrophy, and one had (1.4%) periretinal hemorrhage (Table 1).
2. Demographics of patients
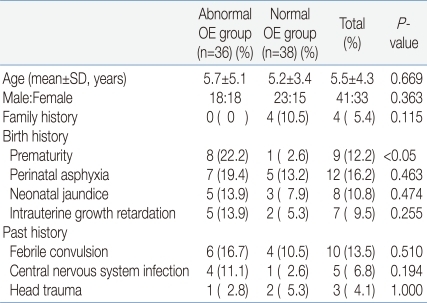
The mean age of the abnormal OE group was 5.7±5.1 years, and that of the normal OE group was 5.2±3.4. The male to female ratio of the abnormal OE group was 1:1 and that of the normal OE group was 1.5:1. There was no significant difference regarding family history for the presence of MD or unexplained early deaths. No significant differences in the frequency of perinatal asphyxia, neonatal jaundice, or intrauterine growth retardation were noted between the two groups. But the history of prematurity was noted in eight patients in the abnormal OE group, significantly higher in prevalence compared to the normal OE group. However, there was no significant difference between the two groups regarding the prevalence of febrile convulsion, central nervous system, and head trauma (Table 2).
3. Clinical manifestations
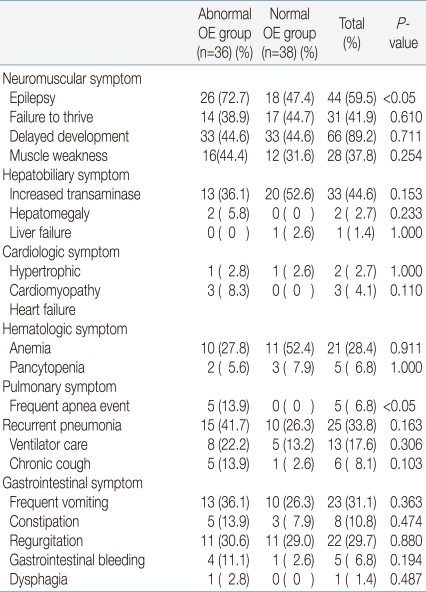
The abnormal OE group had a statistically higher number of patients, twenty-six (72.2%), suffering epilepsy, while the normal OE group had only eighteen (47.4%). There was no significant difference regarding failure to thrive, delayed development, and muscle weaknesses. Hepatobiliary, cardiologic, and hematologic symptoms were also observed in both groups, but without significant difference. Frequent apnea events appeared in five patients (13.9%) in the abnormal OE group, indicating a statistically significant higher prevalence, but there was no other pulmonary symptom of any significant difference. No significant difference in gastrointestinal symptoms was observed between either of the two groups (Table 3).
4. Metabolic screening test, morphological analysis and biochemical enzyme assay
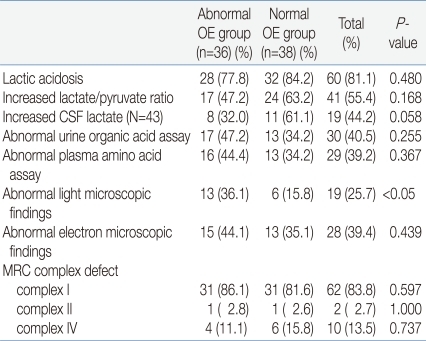
There were twenty-eight (77.8%) patients in the abnormal OE group and thirty-two (84.2%) in the normal OE group who showed the presence of lactic acidosis. Also, seventeen patients (47.2%) of the abnormal OE group and twenty-four patients (63.2%) of the normal OE group showed an increase in their lactate/pyruvate ratio, but with no significant difference. Increased lactate in CSF, abnormal urine organic acids, and abnormal plasma amino acids were observed in both groups, but still with no significant difference. A morphological test was conducted using muscle samples. With a light microscope, abnormal findings were observed in thirteen patients (36.1%) in the abnormal OE group and six patients (15.8%) in the normal OE group implying that there was a statistically significant difference. But with an electron microscope, no statistical difference was noted between the two groups. The enzyme activity of the mitochondrial respiratory chain complex measured by biochemical assay of muscle samples also showed no significant difference between the two groups (Table 4).
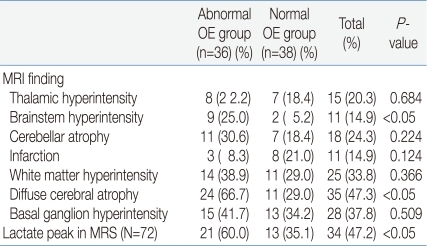
5. Magnetic resonance imaging (MRI) and magnetic resonance spectroscopy (MRS) findings
The MRI findings showed that nine patients (25.0%) in the abnormal OE group and two (5.2%) in the normal OE group had brainstem hyperintensity. Twenty-four patients (66.7%) in the abnormal OE group and eleven patients (29.0%) of the normal OE group had diffuse cerebral atrophy. Both findings were seen statistically more frequent in the abnormal OE group. No other MRI findings showed any significant difference. Among the seventy-two MRS examinees, lactate peaks were observed in twenty-one patients (60.0%) in the abnormal OE group and thirteen patients (35.1%) in the normal OE group. The difference was statistically significant (Table 5).
Discussion
Thus far, there have been many studies regarding the ocular symptoms caused by MD7-10). We also noticed that 50% of the patients in our study suffered from ophthalmologic symptoms and 48.6% actually showed abnormal findings during an OE. In addition, sixteen (21.6%) of the patients with MRC defect but without ophthalmologic symptoms showed abnormal findings in the fundus examination, which emphasizes the importance of an active OE in MD even at the absence of any symptoms. The reason why there are plenty of ocular involvements observed in MD could be explained by the fact that the retinal pigment epithelium possesses a highly metabolically active cell type that has a lot of mitochondria in it, thus susceptible to damages done by mtDNA defects9, 18). Retina pigmentation was the most common pathologic finding noted among our patients during the OE. It is inferred that hypoxic environments due to mtDNA defects cause mitochondrial oxidative stresses and induce reactive oxygen species. This accelerates mtDNA damage which then leads to retinal degeneration18-20).
In this study, we grouped our patients based on their fundus examination results to find out the factors related to ocular involvement in MD. Regarding patients' demographics of age, sex and so forth, there was no particular difference between groups with or without ocular involvement. Prematurity was the only factor among the birth histories that was significantly higher in the abnormal OE group. As for clinical manifestations, the number of frequent apnea events was also significantly higher in the abnormal OE group. It has been reported that frequent apnea events can occur more commonly in patients prematurely born, and hypoxic damage during apnea episodes might have been related with abnormal ophthalmologic findings21). Among the neurological symptoms, epilepsy was the only symptom with significantly higher prevalence in the abnormal OE group, which supports the need for more active OE when epilepsy is present.
It has been reported many times that abnormal findings are observed more frequently under electron microscopy than under light microscopy in cases of pediatric MD4, 16, 22). We also had similar results with our patients and abnormal findings were noticed more often under electron microscopy. But, in our morphological analysis, abnormal findings were observed significantly more often under the light microscopy in the abnormal OE group, but there was no significant difference under the electron microscopy between the two groups. Therefore, it is important that when abnormal findings by the light microscopic examination, such as ragged red fibers, are seen in patients with MD, a more extensive OE should be performed regardless of the symptoms.
Like in other previous studies, diffuse cerebral atrophy was the most frequent finding in the MRI study among our patients23-25). In addition, brainstem hyperintensity was identified significantly more frequently in the abnormal OE group. It is presumed that since ophthalmologic symptoms are likely to be produced through the dysfunction of cranial nerves connected to the brainstem, patients with ophthalmologic symptoms more often showed hyperintensitiy in the corresponding area. In MRS, lactate peaks showed statistically significant correlation to ocular involvement. An increase in CSF lactate rather than serum lactate, and also the detection of lactate peaks in MRS rather than increase in CSF lactate, showed a more significant relationship to ocular involvement. This might have resulted due to tissue specificity2, 17, 18).
However, this study has some limitations. The research was conducted based on relatively small samples because of the rarity of the disease. The scarcity of MD made it difficult to design a prospective study. But if we consider that MD shows a variety of symptoms in several organs and ocular involvement is one of the key symptoms, an OE should always be required, even when the symptoms are not as distinctive. In short, when MD patients have a premature birth history, a history of epilepsy or frequent apnea events, abnormal light microscopic findings in the muscle pathology, diffuse cerebral atrophy in MRI, and brainstem hyperintensity and lactate peaks in MRS, active examinations and diagnoses should always be considered because the possibility of ocular involvement could be higher in such situations. Further prospective studies on more patients are expected in future.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


