


Introduction
Familial hypokalemic periodic paralysis (HOKPP, OMIM no. 170400) is an autosomal
dominant disorder that features reversible attacks of flaccid paralysis with concomitant
hypokalemia1). The first attack can occur any time between infancy and puberty, with the
majority occurring around puberty. Paralytic attacks usually occur at night or early in the
morning and last for hours and sometimes days. They can be induced by carbohydrate- or
sodium-rich meals, strenuous exercise, emotional stress, and exposure to cold2).
The majority of HOKPP cases are caused by mutations in the skeletal muscle voltage-gated calcium channel gene, CACNA1S, or in the sodium channel gene, SCN4A1). These mutations have been shown to contribute to an inward cation leakage current (referred to as the gating-pore current), which makes the muscle fibers of patients susceptible to abnormal depolarization in response to low extracellular potassium concentration3,4). Alterations in the expression, subcellular localization, and/or kinetics of nonmutated potassium channels, which reduce outward potassium currents, have been implicated in the development of hypokalemia as well as pathological depolarization5,6,7,8). However, the reason why potassium channels are affected by mutations in the CACNA1S or SCN4A gene is not yet clear.
A previous study demonstrated that skeletal muscle fibers from HOKPP patients exhibited increased intracellular calcium levels compared with normal cells6). An increase in cytoplasmic calcium levels via its entry and/or release from intracellular stores triggers a variety of physiological processes, such as muscle contraction, neuronal excitability, vasoregulation, hormone secretion, and immune responses9,10,11,12). The elevated intracellular calcium ions, however, also stimulate the activity of calcium-activated potassium (KCa) channels, of which the large-conductance KCa channels (also termed big potassium [BK] channels) primarily serve as negative feedback regulators that repolarize the cell by increasing potassium efflux. This can lead to membrane hyperpolarization and a consequent decrease in cell excitability13).
We conducted this study to test the hypothesis that the expression patterns of the KCa channel genes (KCNMA1, KCNN1, KCNN2, KCNN3, and KCNN4) in the skeletal muscle cells of HOKPP patients differ from those in normal cells and to find a mechanistic link between HOKPP mutant ion channels and pathogenic alterations to non-mutant potassium channels.
Materials and methods
1. Subjects
We reviewed the records of 185 patients who were being treated for HOKPP from March 2008 to April 2013 in the clinics of Seoul Children's Hospital and Konyang University Hospital. Each patient underwent genetic testing. All the patients fulfilled the diagnostic criteria for HOKPP, including (1) episodic attacks of muscle weakness with hypokalemia (<3.5 mEq/L); (2) positive family history or genetically confirmed skeletal calcium or sodium channel mutation; and (3) exclusion of secondary causes of hypokalemia, such as thyroid, adrenal, and renal disorders, and drug abuse2,14). Mutation screening was performed by sequencing the entire coding region of the CACNA1S, SCN4A, and KCNJ2 genes, as described elsewhere15,16). The clinical details of some patients have been reported previously2,16,17,18). Of these, we selected the three who presented with the most obvious symptoms of HOKPP in terms of both severity and frequency of paralytic attacks. These patients had the Arg1239Gly mutation in the CACNA1S gene. Three healthy volunteers acted as controls. All participants provided written informed consent, and the study was conducted in compliance with the guidelines of the Institutional Review Board of Konyang University Hospital.
2. Methods
1) Skeletal muscle tissue sampling and preparation
Skeletal muscle biopsy samples were obtained as described previously7). The muscle biopsy samples were rinsed in phosphate buffered saline (PBS) with Ca2+-Mg2+ and dissected into small pieces (diameter, 1-2 mm) with a sterilized scalpel. The tissue fragments were cultured in Dulbecco's modified Eagle's Medium (DMEM; Thermo Scientific, Marietta, OH, USA) supplemented with 5% fetal bovine serum (FBS; Thermo Scientific), 2µM glutamine (Sigma-Aldrich, St. Louis, MO, USA), 1% nonessential amino acids (Thermo Scientific), 0.1mM β-mercaptoethanol (Sigma-Aldrich), 5-ng/mL fibroblast growth factor-basic recombinant human protein (Invitrogen, Carlsbad, CA, USA), and 1% penicillin-streptomycin (Caisson, Logan, UT, USA) for 2 weeks at 37℃ in an incubator containing 95% air and 5% CO2 (Thermo Scientific). Isolation of skeletal muscle cells from the original culture was performed according to a method described previously19).
2) Preparation of potassium buffers
Potassium buffer (4mM) at pH 7.35 (4mM KCl, 145mM NaCl, 1mM MgCl2, 0.5mM CaCl2, 5mM glucose, and 10mM 3-(N-morpholino) propanesulfonic acid [MOPS]) was prepared and used for exposing cells to a normal physiological concentration of extracellular potassium. To induce depolarization of skeletal muscle cells via a high concentration of extracellular potassium, 50 mM potassium buffer (50 mM KCl, 145 mM NaCl, 1 mM MgCl2, 0.5 mM CaCl2, 5 mM glucose, and 10 mM MOPS; pH 7.35) was prepared. The buffers were sterilized before use.
3) Culturing of skeletal muscle cells and treatment with potassium buffer solutions
Skeletal muscle cells obtained from HOKPP patients (patient cells) and healthy controls (normal cells) were cultured in DMEM containing 20% FBS (Thermo Scientific) and 1% penicillin-streptomycin at 37℃ in an incubator containing 95% air and 5% CO2. To induce differentiation, skeletal muscle cells were incubated in DMEM supplemented with 2% horse serum (Thermo Scientific) and 1% penicillin-streptomycin for up to 5 days. The differentiation medium was replaced every 48 hours. Both normal and patient cells were collected for analysis during the 12th passage. The mRNA and protein levels of the KCa channel genes (KCNMA1, KCNN1, KCNN2, KCNN3, and KCNN4) were analyzed in both types of cells, prior to and at 1 hour after exposure to the 4 and 50 mM potassium buffers.
4) Measurement of cytosolic calcium levels
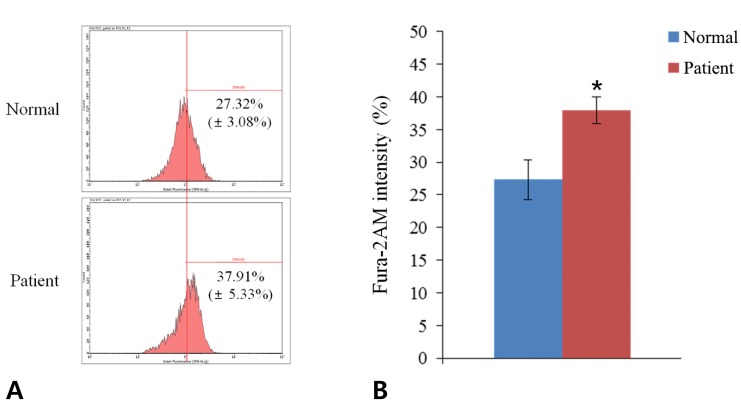
Cytosolic calcium levels were analyzed using fura-2-acetoxymethyl ester (Fura-2AM), a membrane-permeable, calcium-sensitive fluorescent dye (Sigma-Aldrich). Both normal and patient cells were loaded with 1µM Fura-2AM diluted in 4mM potassium buffer for 30 minutes in a CO2 incubator. After washing with potassium buffer to remove residual dye, the cells were treated with trypsin-ethylenediaminetetraacetic acid (Caisson). They were then harvested, washed with Ca2+-free PBS, and analyzed by flow cytometry (Millipore, Billerica, MA, USA). All the experiment protocols were done in triplicate.
5) Quantitative reverse transcription polymerase chain reaction analysis
Total RNA from the skeletal muscle cells was extracted using TRIzol reagent (Invitrogen), and 100 ng was converted to cDNA by using SuperScript III reverse transcriptase (Invitrogen) according to the manufacturer's instructions. AccuPower PCR PreMix (Bioneer, Daejun, Korea) was added to the reaction mix, and quantitative reverse transcription polymerase chain reaction (RT-PCR) analysis was performed using primers for KCNMA1, KCNN1, KCNN2, KCNN3, and KCNN4. Primers were designed with the VectorNTI10 (Invitrogen) and Primer3 software (http://sourceforge.net/projects/primer3/). The sequences of these primers are available on request. The PCR conditions consisted of initial denaturation at 95℃ for 5 minutes, followed by 30 cycles at 95℃ for 30 seconds, 55℃ for 30 seconds, and 72℃ for 1 minute, with final extension at 72℃ for 5 minutes. All the reactions were performed in triplicate. The mRNA expression levels for each gene were normalized to the expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
6) Western blot analysis
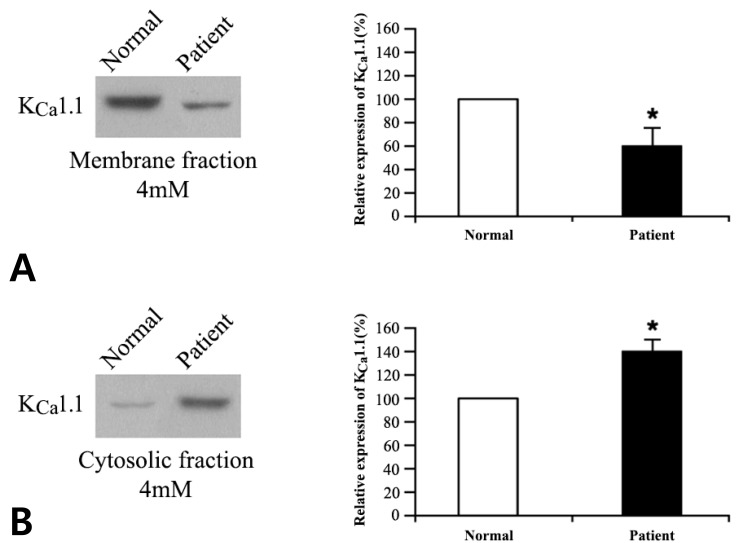
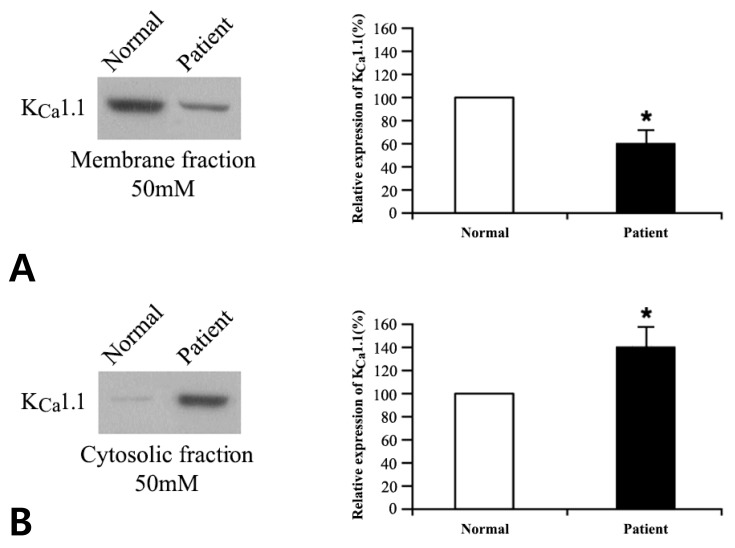
Prior to and after exposure to the two different concentrations of potassium buffers for 1 hour, the cytosolic and membranous protein fractions of normal and patient cells were separated and analyzed by western blotting. Detailed methods for subcellular fractionation and western blot analysis have been previously described8). To detect the channel proteins, primary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for KCa1.1 (MaxiKα [B-1]), KCa2.1 (SK1 [A-13]), KCa2.2 (KCNN2 [U-24]), KCa2.3 (SK3 [H-45]), KCa3.1 (IK1 [D-5]), and β-actin (β-actin [C4]) were used in combination with antigoat, antimouse, or antirabbit secondary antibodies. The proteins were then quantified using the TINA 2.0 densitometric analytical system (Raytest Isotopenmeβgeräte GmbH, Straubenhardt, Germany) according to the manufacturer's instructions. All the experiments were performed independently at least three times.
Results
1. Cytosolic calcium levels in skeletal muscle cells
We examined cytosolic calcium levels in both patient and normal cells in 4mM potassium buffer using the calcium-sensitive dye Fura-2AM. The percentage of fluorescence was measured by flow cytometry. At least 5,000 cells were analyzed for each sample by using InCite software ver. 2.2.2 (Millipore), and the threshold was matched at SSC 291. Patient cells exhibited significantly higher levels of cytosolic calcium ions than normal cells (37.91%±5.33% vs. 27.32%±3.08%, P<0.05) (Fig. 1).
2. KCa channel mRNA expression
We performed quantitative RT-PCR to compare the mRNA levels for the KCa channel genes (KCNMA1, KCNN1, KCNN2, KCNN3, and KCNN4) between patient and normal (control) cells in both 4mM and 50mM potassium buffers. No significant differences were found at either potassium concentration (data not shown).
3. Western blot analysis to evaluate the expression patterns of KCa channel proteins
We compared the protein expression profiles of the KCa channels in patient versus normal cells, following exposure to 4 and 50mM potassium. Membranous and cytosolic fractions of the cells were separated, and the protein expression levels of the KCa channel genes were evaluated by western blot as mentioned above. No significant difference was observed for KCa2.1, KCa2.2, KCa2.3, and KCa3.1 (data not shown); however, the expression of KCa1.1 (encoded by KCNMA1) differed significantly in patient cells: compared with control levels, expression was lower in the membrane fraction (P<0.05) and higher in the cytosolic fraction (P<0.05) for both 4 and 50 mM potassium buffers (Figs. 2, 3).
Discussion
Previous studies have reported alterations in the expression, subcellular localization, and/or kinetics of nonmutant potassium channels (but not BK channels) in HOKPP5,6,7,8). However, the relationship between mutations in the CACNA1S or SCN4A gene and the pathological operation of nonmutant potassium channels has not yet been elucidated.
The mutant channels responsible for HOKPP have been shown to generate a nonselective inward cation leakage current through an aberrant gating pore that is open at the resting membrane potential3,4). In agreement with the results of Puwanant and Ruff6), we observed an increase in the cytoplasmic calcium levels of HOKPP patient muscle cells at a normal physiological concentration of extracellular potassium. Although the precise mechanism responsible for this increase is not known, from the findings of the above studies3,4), we can reasonably infer that it occurs via inward calcium leakage through mutant channels and/or calcium release from intracellular stores in response to an inward cation current through the mutant channels at resting potential.
The BK channel exists as a tetrameric structure, composed of four α-subunits, either alone or associated with β-subunits. The α-subunit, encoded by KCNMA1, is the pore-forming unit, and the β-subunit, encoded by KCNMB1, KCNMB2, KCNMB3, or KCNMB4, is the modulatory unit. The different subunit compositions of BK channels give rise to its functional diversity in various tissues; for example, skeletal muscle BK is assembled from α-subunits alone, whereas vascular BK is composed of α+β1 and neuronal types are composed of α+β4 or α+β220,21,22). Additional mechanisms involving multiple promoters, alternative splicing, and metabolic regulation also modify BK functional properties, thus producing the diverse range of phenotypes necessary for the normal functioning of many tissues23,24,25,26). Consistent with the wide distribution of BK channels throughout the human body, malfunction of BK channels has been implicated in various medical conditions, such as epilepsy, psychiatric disorders, deafness, hypertension, asthma, and urinary incontinence10,27,28,29,30,31). BK channels play an essential role in regulating both the firing frequency and the action potential repolarization phase of muscle cells. BK channels also serve as molecular targets for drugs used in the treatment of HOKPP: acetazolamide and dichlorphenamide, which act by opening skeletal muscle BK channels32).
This is the first study to demonstrate altered subcellular distribution of BK channels in the skeletal muscle cells of patients with HOKPP. Paralysis in HOKPP results from persistent membrane depolarization, and skeletal muscle fibers from patients with HOKPP are extremely susceptible to depolarization-induced inactivation33). Given the pivotal role played by BK channels in action potential repolarization by increasing potassium efflux out of cells, the reduced membrane expression of BK channels in patient cells, observed in this study, correlates well with the characteristic pathological conditions of HOKPP, such as hypokalemia and paralysis.
The altered subcellular distribution of BK channels in patient cells in the 4 mM potassium buffer may represent a physiological mechanism developed to compensate for the chronic abnormal increase in cytoplasmic calcium at resting potential. Indeed, several posttranslational modifications have been reported to affect the membrane expression of BK channels25,26). On the other hand, it is currently unclear how the altered subcellular distribution of BK channels is maintained in patient cells in the depolarizing 50 mM potassium buffer. Whether alternative splicing of the KCNMA1 gene or certain posttranslational mechanisms modify the subcellular distribution of BK channels in patient cells remains to be determined.
In conclusion, we observed an abnormal increase in cytoplasmic calcium levels and altered subcellular distribution of BK channels in the skeletal muscle cells of patients with HOKPP. The altered distribution of BK channels represents a novel mechanism linking elevated intracellular calcium, induced by HOKPP-associated mutations, to the hypokalemia and paralysis that is symptomatic of the disease and also demonstrates a connection between HOKPP mutant ion channels and pathogenic changes in nonmutant potassium channels.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


