


Introduction
Fanconi anemia (FA) is a rare autosomal and X-linked recessive disorder, characterized by physical abnormalities, progressive bone marrow failure (BMF), hypersensitivity to DNA cross-linking agents and predisposition to malignancy1). It was first described in 1927 by Fanconi2), who described a family in which three children had pancytopenia and birth defects, such as short stature, hypogonadism and skin disorder. Since then, 1,075 patients were registered in the International Fanconi Anemia Registry (IFAR) between May 1982 and August 20083).
FA is considered the most common inherited cause of BMF, which is usually apparent between 6 and 8 years of age. Most patients with FA have birth defects including skin hyperpigmentation and/or cafe au lait spots, skeletal malformations, short stature and urogenital abnormalities3). However, one-third of the patients have few or none of these features, which makes it difficult to diagnose4). FA patients may develop myelodysplastic syndrome (MDS), or acute myeloid leukemia (AML), with relative risk of developing AML being 800-fold higher than that of the general population. There is also a strong predisposition to specific solid tumors, including head, neck, gynecological squamous cell carcinoma (SCC) and liver tumors4,5).
Hematopoietic stem cell transplantation (SCT) is currently the only treatment to restore normal hematopoiesis because of nonresponse to immunosuppressive therapy, such as antilymphocyte globulin, antithymocyte globulin (ATG) and cyclosporine, which is usually given to treat idiopathic aplastic anemia (AA)6). Moreover, because of hypersensitivity to chemotherapeutic agents or radiation, higher risk of developing malignancies, and potential recurrence in the siblings, the approach to SCT should be different from that of idiopathic AA in terms of the donor selection, optimal timing of SCT, and the choice of conditioning regimen. Also, the prognosis after SCT is quite different7,8). Therefore, FA should be sought carefully in any patients with AA or BMF syndromes in children and young adults.
A screening study with DNA cross-linking agents in patients with BMF and clinical manifestation of 6 FA patients was published from our institution in 1997 and 1998, respectively9,10). The present study represents an update and long-term follow-up study of 12 patients diagnosed with FA at the Chonnam National University Hospital over the last 20 years. Clinical presentations, laboratory findings, diagnostic methods, treatment modalities, outcomes and long-term sequelae of patients were retrospectively analyzed to increase awareness of this rare but very challenging genetic disease.
Materials and methods
1. Subjects
This study included 12 FA patients seen at the Department of Pediatrics, Chonnam National University Hospital and Chonnam National University Hwasun Hospital between January 1991 and December 2012. The patients were diagnosed on the basis of family history, physical findings in addition to hematologic abnormalities, and chromosomal breakage tests induced by diepoxybutane (DEB) or mitomycin-C (MMC)3,4).
2. Chromosome breakage tests
The screening for the diagnosis of FA chromosome breakage tests was performed on 104 subjects including 78 AA, 6 MDS, 6 acute leukemia, 2 idiopathic thrombocytopenic purpura, 5 Diamond-Blackfan anemia, and 7 sibling of FA between January 1996 to December 2012.
Chromosome breakage tests with DEB and MMC were carried out according to the technique described by Auerbach et al.11) with minor modifications10). Two patients (cases 11, 12) with negative blood results underwent chromosomal breakage tests on cultured skin fibroblasts. Gene analysis for FANCA was performed in case 12 using a multiplex ligation-dependent probe amplification (MLPA). The gene dosage assay for FANCA to detect deletion/duplications was carried out using a SALSA MLPA Kit P031-P032 FANCA (MRC-Holland, Amsterdam, the Netherlands) according to the manufacturer's instructions.
3. Retrospective analysis
A retrospective analysis of the patients' clinical manifestations, diagnostic methods, treatment modalities, outcomes and long-term sequelae was performed. Height and weight were standardized for age and gender using data from the Korea Center for Disease Control and Prevention (2007)12). Short stature was designated when height was lower than 2 standard deviation score.
The following were the criteria for the response to the treatment. Complete response was defined when hemoglobin level ≥age-adjusted level, and absolute neutrophil count (ANC) ≥1.5×109/L, and platelet count ≥100×109/L. Partial response was defined when there is no requirement for red cell transfusion or reticulocyte count increase ≥30×109/L, ANC increase ≥0.3×109/L, and no platelet transfusion and platelet increase ≥20×109/L13).
Survival was assessed on the date of last patient contact and analyzed as of December 2012. Statistical analysis was performed using SPSS ver. 19.0 (SAS Institute Inc., Chicago, IL, USA). The probability of overall survival (OS) was estimated according to the Kaplan-Meier (K-M) survival method. Failure-free survival (FFS) was defined as survival with response. Death, transfusion-dependency, disease progression to MDS or AML, a second course of SCT, and relapse were considered treatment failures14). Statistical significance was defined as P<0.05.
Results
1. Clinical manifestations
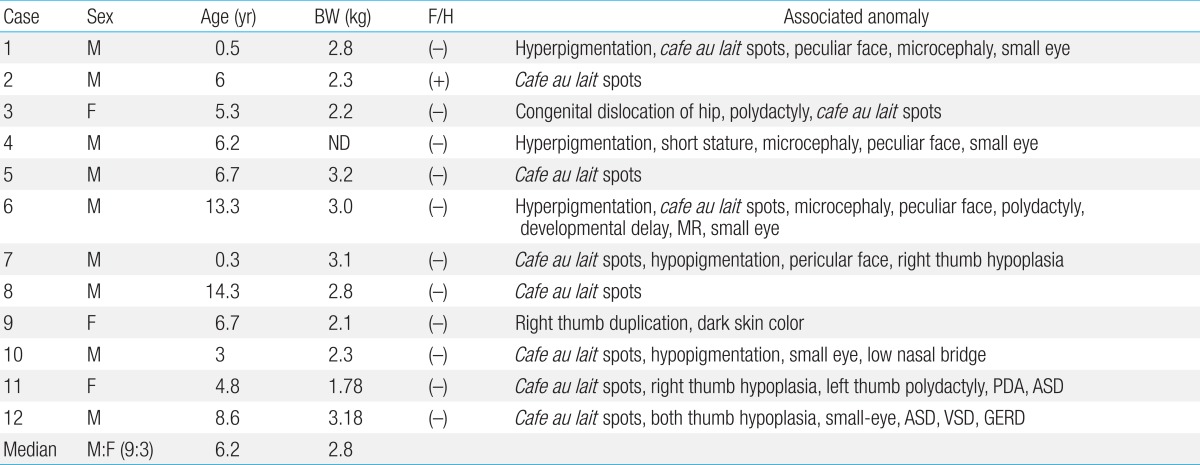
Of 12 patients with a diagnosis of FA, 9 were males and 3 were females. Their age at first evaluation ranged from 5 months to 14.3 years (median, 6.2 years). Characteristics of the patients are summarized in Table 1.
One patient (case 2) had a family history in which his elder brother died of AA. Low birth weight was observed in 7 patients (63.6%), with the median body weight of 2.8 kg. All patients had at least one of the following associated anomalies: skin pigmentation including cafe au lait spots, hyperpigmentation or hypopigmentation (100%), skeletal abnormality (50.0%), microophthalmia (33.3%), cardiac anomaly (16.7%), microcephaly (16.7%), and short stature (8.3%). Other findings included developmental delay, congenital dislocation of hip, and polydactyly (Table 2).
2. Hematologic findings
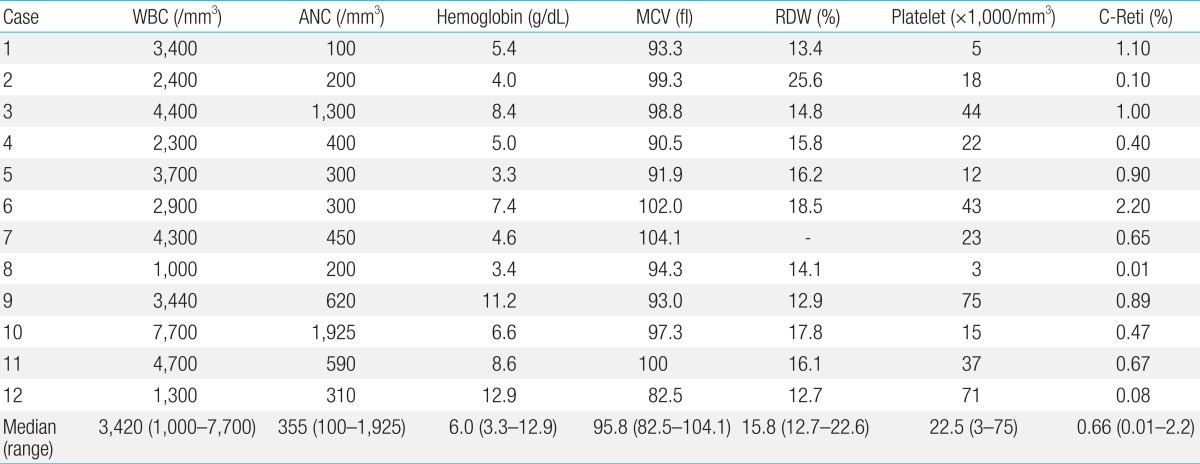
Laboratory findings of the patients at diagnosis are summarized in Table 3. All patients had evidence of BMF at the time of their first visit. The median blood counts were: white blood cell, 3,420/mm3; hemoglobin, 6.0 g/dL; and platelets, 22,500/mm3. Most patients had macrocytosis with median mean corpuscular volume of 95.8 fL. A bone marrow exam showed hypocellular marrow with loss of hematopoietic cells, and fatty replacement in all patients except one. The 3-month-old patient (case 7) had a normocellular marrow with macrocytosis, erythroid hyperplasia and significant dyserythropoiesis which was compatible with MDS15). According to the classification of AA by severity16), 7 patients (63.6%; cases 1, 2, 4, 5, 8, 10, and 12) among 11 patients diagnosed as AA had severe AA including 2 very severe diseases (cases 1, 2).
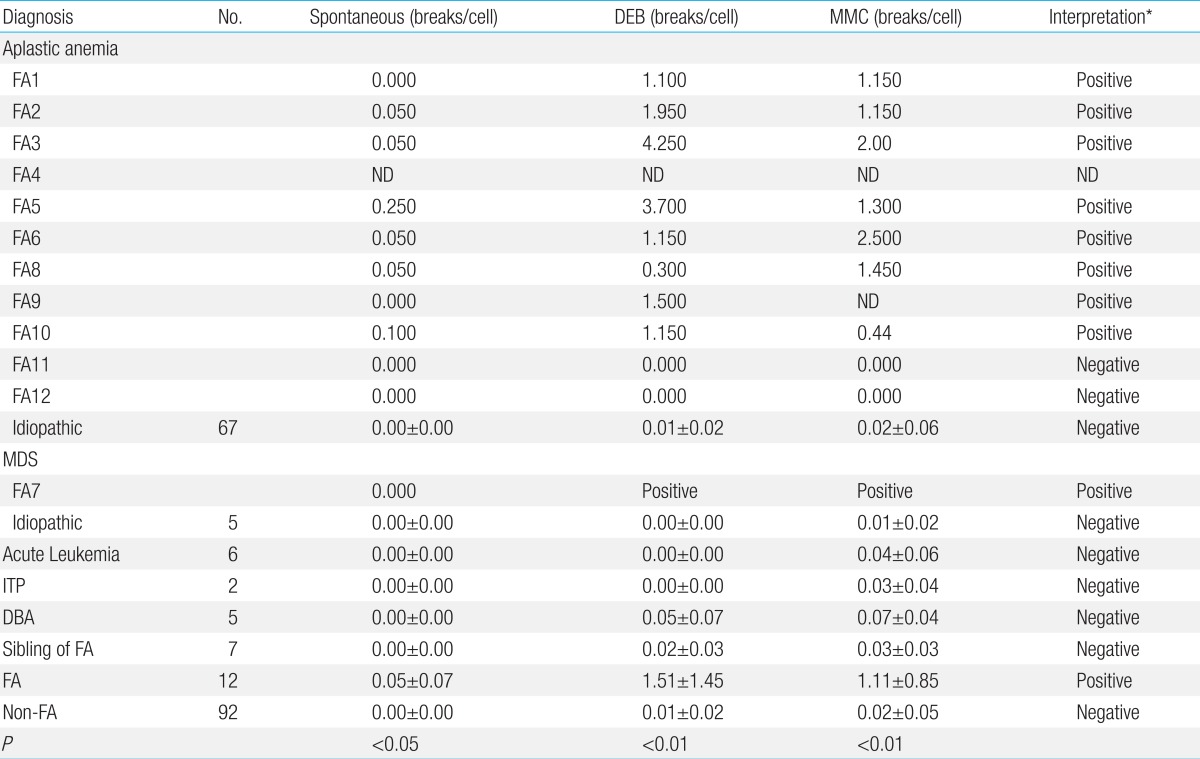
3. Chromosomal breakage test
Chromosomal breakage tests with DEB and MMC have successfully discriminated the two groups among 104 subjects: FA patients with hypersensitivity to both DEB and MMC, and non-FA patients with no hypersensitivity. The FA patients showed increased chromosomal breaks/cell of 1.51±1.45 (mean±standard deviation) (range, 0 to 4.25) to DEB (P<0.01), and 1.11±0.85 (range, 0 to 2.5) to MMC (P<0.01) (Table 4). Nine cases were found to have increased chromosomal breakage to DEB and MMC, confirming the diagnosis of FA. The remaining 3 patients (cases 4, 11, and 12) were diagnosed on the basis of characteristic physical stigma in addition to hematologic abnormalities. Case 4 died before the availability of the tests. Breakage tests on skin fibroblasts failed unfortunately in case 11 and were negative in case 12. FANC-A mutation was not detected in case 12.
Ninety-two subjects with diseases other than FA including 67 patients with idiopathic AA showed no increased hypersensitivity: chromosomal breakages/cell of 0.01±0.02 (range, 0 to 0.1) to DEB and 0.02±0.05 (range, 0 to 0.4) to MMC, respectively. No false positivity was found among subjects other than FA. Increased spontaneous breakage was found in 6 among 11 FA patients, and thus was not sensitive as a diagnostic method.
4. Treatment
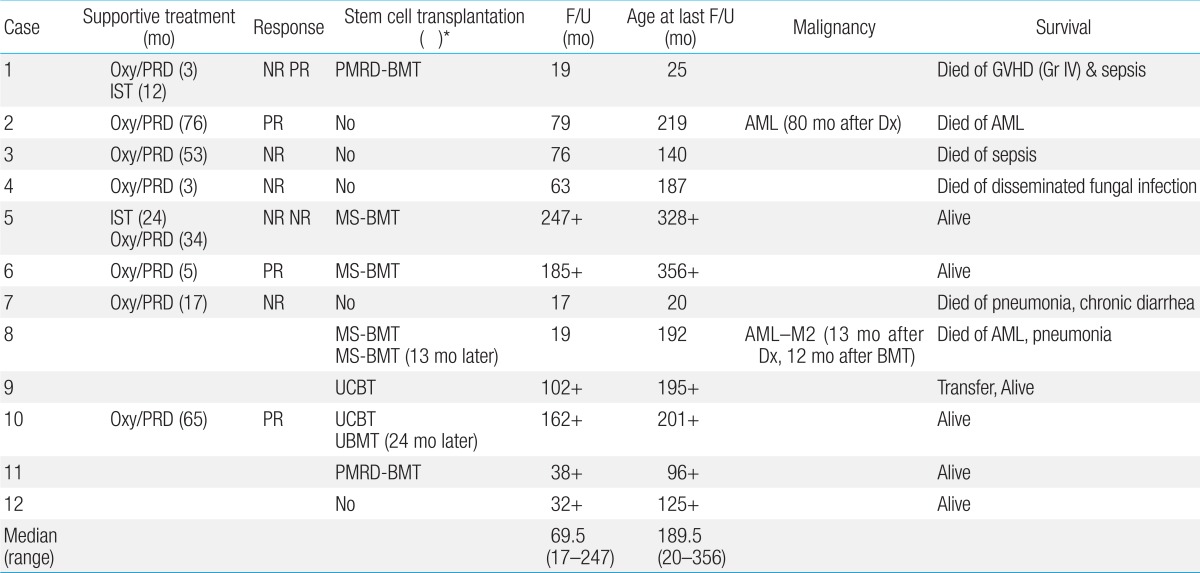
Even though the treatment modalities varied with time, FA patients received a combination of supportive treatment, oxymetholone, prednisolone, immunosuppressive treatment with ATG, and SCT (Table 5). In earlier years, 8 patients were treated with oxymetholone and prednisolone initially for 3 to 76 months. Oxymetholone and prednisolone treatment was partially beneficial in 3 patients (37.5%) (cases 2, 6, and 10), although peliosis hepatis (case 10) and masculinization (case 3) developed as side effects. All 4 patients (cases 2, 3, 4, and 7) treated with oxymetholone and prednisolone alone eventually died of invasive fungal infection, pneumonia, sepsis and development of AML, respectively.
Two patients received immunosuppressive treatment. Case 5 was initially diagnosed with idiopathic AA before the availability of chromosomal breakage test as he showed minimal physical stigma. He was later diagnosed with FA and transplanted. Case 1 with severe disease had received oxymetholone, prednisolone and ATG treatment to no avail before he underwent a mismatched SCT.
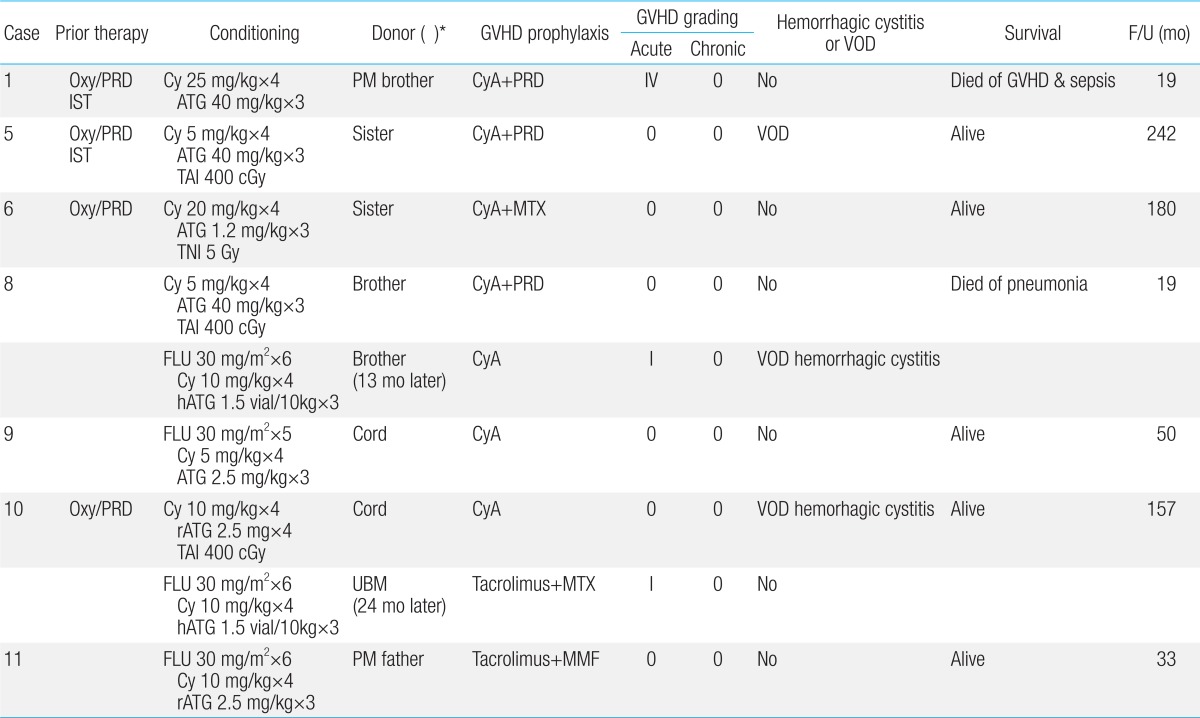
Seven patients underwent 9 hematopoietic SCTs. The median age at transplant was 11.7 years. The stem cell sources were: matched siblings (n=4), partially matched related donors (n=2), unrelated cord blood (n=2), and unrelated bone marrow (n=1). Case 8 showed initial graft rejection after matched sibling transplant, and underwent a second transplant from the same donor when he developed AML 1 year later. Case 10 suffered from engraftment failure after initial cord blood transplant (CBT), but was rescued by unrelated bone marrow. Another patient (case 9) with CBT also rejected initial graft, but she showed autologous recovery. None was T cell depleted.
The conditioning regimens varied over 2 decades. In earlier years, low-dose cyclophosphamide (Cy), ATG with or without low-dose irradiation were mainly used (n=5; Table 6). Recently, fludaradine (Flu)-based containing regimen without irradiation was used (n=4).
5. Survival
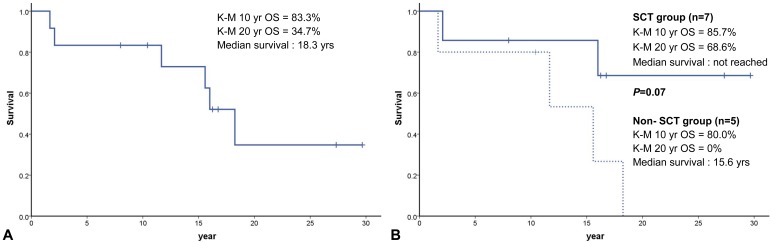
Among 12 FA patients with the median age of 189.5 months (range, 20 to 356 months) at last follow-up, six patients (50%) are alive at a median follow-up of 69.5 months (range, 17 to 247 months) with the median age of 198 months (range, 96 to 356 months) at last follow-up. The K-M probability of OS was 83.3% at 10 years and 34.7% at 20 years for the entire group. Median survival was 18.3 years (range, 14.9 to 21.6 years) after diagnosis (Fig. 1A). The median survival of non-SCT group was 15.6 years (range, 4.0 to 27.1 years), while that of the SCT group was not reached (P=0.07). For the SCT group, the K-M probability of survival after diagnosis was 85.7% at 10 years and 68.6% at 20 years, respectively (Fig. 1B).
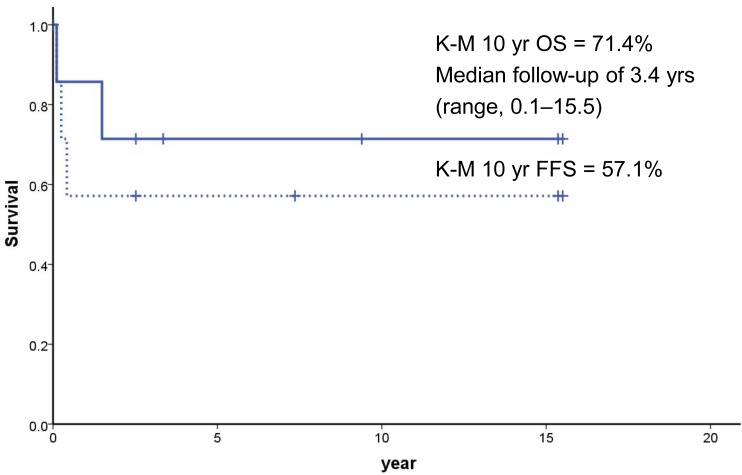
Among 7 who underwent SCTs, 5 are alive with the K-M 10-year OS of 71.4% after SCT with the median follow-up of 3.4 years. The K-M probability of FFS at 10 years was 57.1% after SCT (Fig. 2). Case 1 who underwent mismatched related donor transplant died of grade IV acute graft-versus-host disease (GVHD) at 40 days after transplant, while another patient (case 8) who developed AML (M2) died at 12 months after initial transplant. Because of the small number of cases, the survival was not different by the conditioning method no matter whether the patients received Cy-based, Flu-based, or radiation containing regimens. Also, the use of oxymetholone and prednisolone prior to SCT was not associated with poor outcome as 3 of 4 patients (cases 1, 5, 6, and 10) are surviving.
Infection was the main cause of deaths, as seen in 5 patients (83.3%). Acute leukemia developed in 2 patients (cases 2 and 8), while solid tumors have thus far not been identified.
6. Late sequelae
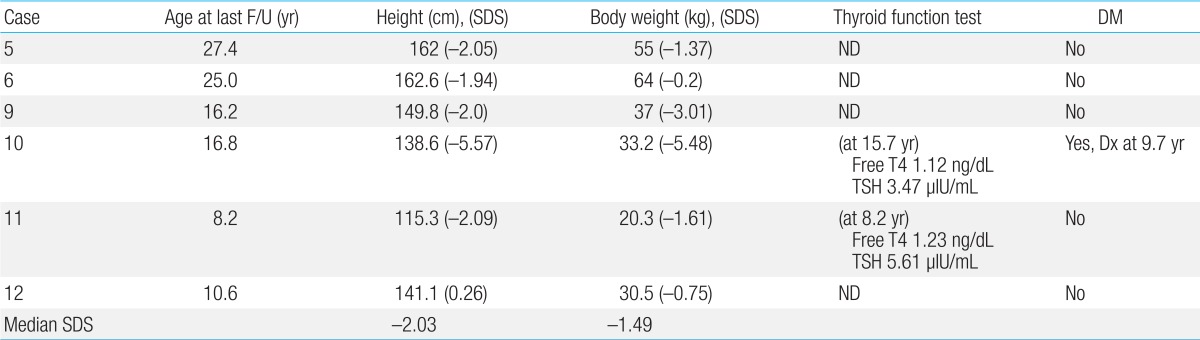
Among the 6 patients who are alive, 3 patients are suffering from short stature with less than 2 standard deviation score in height (Table 7). The median standard deviation score was -2.03 in height and -1.49 in body weight, respectively. No surviving patient showed symptoms of hypothyroidism and 2 patients evaluated for thyroid function revealed normal values. Case 10, who received huge amounts of transfusions after the initial graft rejection, developed an insulin-dependent diabetes mellitus secondary to iron accumulation in the pancreas with elevated ferritin level of 5,961 ng/mL, requiring insulin treatment.
Discussion
FA is a genetic disorder characterized by multiple congenital anomalies, hematological abnormalities and predisposition to a variety of cancers. Approximately two-thirds of FA patients present with a combination of various congenital abnormalities such as short stature, Fanconi facies with microophthalmia, skeletal deformities, skin hyperpigmentation, and cardiac, renal, genitourinary, and/or other malformations3,17). Skin abnormalities have been described in the literature in 55% of cases of FA4). However, all patients in this study had skin hyperpigmentation and/or cafe-au-lait spots. Skeletal abnormalities, found in 43% of cases of FA, were hypoplasia or absence of the thumbs, bifid or supernumerary thumbs, and hypoplasia or absence of the radius4). One third in this study had skeletal abnormalities. Other less common abnormalities were 2 cardiac anomalies and 1 gastrointestinal defect. Physical abnormalities can be subtle or absent as 3 patients (cases 2, 5, and 8) in the present study showed cafe-au-lait spots only.
Hematologic abnormalities are the most important clinical features of FA. BMF develops at a median age of 7 years (range, birth to 41 years)5,18). For the majority of patients, the suspicion of FA is made only after the onset of pancytopenia. Initial hematologic findings are diverse. At birth, blood count is usually normal and macrocytosis is commonly the first detected abnormality followed by thrombocytopenia and anemia. During the first decade of life, about 75%-90% of FA patients develop BMF18).
In some patients, the underlying diagnosis of FA is not known until MDS or AML occurs. The relative risk of developing AML is approximately 800 fold higher than that of the general population, with the median onset of 14 years19). In this study, 2 patients (cases 2 and 8) developed AML at 15.4 and 18.1 years of age, respectively. MDS was detected in 23 cases among 119 FA patients cohort at a median of 12 years (range, 2 to 44 years) in a recent study20). The presentation of MDS at the age of 3 months (case 3) in this study should be extremely rare15).
In addition to hematologic malignancy, solid tumors develop at markedly higher rates in FA patients. Most of the solid tumors in FA patients are SCC, particularly of the head and neck, esophagus and anogenital regions. Hepatic tumors may also be found at higher rates, which may be related to androgen use. Approximately one-third of FA patients were known to develop a solid tumor by the fourth decade of life21). However, not a single case of solid tumor has thus far been identified in this study. With wider application of SCT and improved survival, long-term survivors will have a particularly high risk of developing solid tumors.
Because of the heterogeneity on clinical features and recessive mode of inheritance FA can not be diagnosed with certainty by careful history and physical examination in patients with BMF. The biologic diagnosis of FA is primarily based on the exquisite sensitivity of FA cells to the cytotoxic and clastogenic effect of DNA cross-linking agents such as DEB or MMC. The chromosomal breakage test with these agents is the technique of reference for diagnosing FA3,10,11). The incidence of FA has been reported to represent 25% to 30% of pediatric AA. In Korea, however, the frequency of FA among patients with AA has not been systematically evaluated and has probably been underestimated as DEB and MMC tests have not been routinely incorporated in the evaluation of patients with AA in every institution. In accordance with the previous study10), the chromosomal breakage of FA cells to DEB and MMC was distinguishable from non-FA group in this study. Spontaneous chromosomal breakage was not effective in distinguishing FA from non-FA patients because of overlap of breakage ranges11). In the current study, 5 FA patients did not show spontaneous chromosomal breakages.
Other blood tests such as cell-cycle analysis and evaluation of FANCD2 mono-ubiquitination may also be used to diagnose FA. However, all of these tests can be ambiguous or even falsely negative in patients who develop somatic mosaicism and hematopoietic reversion22). Because these phenomena have not been demonstrated in fibroblasts, primary fibroblast cells can be tested for chromosomal breakage tests or other tests for FA diagnosis23). In the current study, 2 patients (cases 11 and 12) with peculiar physical characteristics along with BMF showed negative results for chromosomal breakage tests on blood. Attempts on skin fibroblasts were unfortunately not successful in case 11 and negative in case 12. The possibility of somatic mosaicism or hematopoietic reversion should be contemplated in those cases.
Since the first FA gene was demonstrated in 199224), 16 FA complementation groups (FANCs) have been identified (FANC-A, -B, -C, -D1, -D2, -E, -F, -G, -I, -J, -L, -M, -N, -O, -P, and -Q) so far. The majority of FA genes are located on autosomes except FANCB, which is on the X chromosome. Among the 15 complementation groups, FA-A (60%-70%), FA-C (up to 14%), and FA-G (up to 10%) collectively accounts for more than 90% of the FA groups in the western population17). However, considerable variation in the frequency of each complementation group has been reported according to ethnicity. FA-A and FA-G mutations were detected in 70%-80% and 10%-22% in Japanese population25). A recent study from Korea identified 6 FA-A (46%), 7 FA-G (54%) and no FA-C among 13 FA patients using MLPA and direct sequencing26). Unfortunately, 1 patient (case 12) who underwent gene analysis was not able to be subtyped in the present study.
It is crucial to identify patients with FA in the management of BMF patients. Patients with BMF who happen to have underlying undiagnosed FA will not respond to the immunosuppressive therapy that is used to treat patients with idopathic AA. Moreover, patients with FA will often die of toxicity due to hypersensitivity to chemoradiotherapy when given conventional conditioning for SCT, and therefore reduced intensity conditioning regimens should be used for FA patients8). Also, human leukocyte antigen (HLA)-matched sibling donors should also be screened for FA as the recurrence rate for FA in a sibling is 25% for autosomal recessive disease.
In FA patients, hematological complications are the most life-threatening event. According to the IFAR report, the risk of developing BMF and hematological malignancies by 40 years is 90% and 33%, respectively5). Hematopoietic SCT remains the only treatment to correct the hematologic manifestations in FA patients. Because of high sensitivity to radiation and conditioning agents (alkylating agent), alternative conditioning regimens using lower dose Cy, using Flu and avoiding irradiation have shown hopeful results so far27).
Recently, a promising outcome using Flu (120-180 mg/m2), low-dose Cy (40 mg/kg) and ATG based regimens was reported in 8 FA patients. Donors were either related (n=4), or unrelated (n=4). All patients achieved hematopoietic recovery and were alive at median follow up of 72 months (range, 4 to 117 months)28). In the United Kingdom, a similar nonradiation conditioning regimen consisting of Flu 125-150 mg/m2, Cy 20-30 mg/kg and ATG was used for 7 FA patients. They were transplanted from unrelated umbilical cord blood (UCB) (n=4), HLA matched unrelated (n=2) and haploidentical (n=1) peripheral blood grafts. All patients were alive at median follow-up of 37 months (range, 13 to 54 months), although 2 rejected their grafts initially, but were rescued by a second transplant7). In the present study, however, the survival was not different according to the conditioning method possibly secondary to the small number of cases.
SCT from HLA-matched sibling donor generally gives the best outcome, if performed early prior to the development of MDS or leukemia27). The data from the International Bone Marrow Transplant Registry showed that, among 209 patients transplanted from matched siblings between 1994 and 1999, the 3-year survival was 81% in patients <10 years of age (n=109) and 69% in older patients (n=100)29).
However, the majority of FA patients do not have a HLA matched sibling donor. Until recently, SCT from an alternative donor (i.e., unrelated or HLA mismatched related) has been markedly less successful due to the high rate of graft failure, regimen related toxicity, GVHD and opportunistic infections27). In the present study, among 2 transplanted from partially matched related donors case 1 died of grade IV acute GVHD and sepsis.
Since the first successful transplant in 1989 for a FA patient30), UCB has been increasingly used as a donor source. Although the engraftment after UCB transplant (UCBT) was slower, engraftment and survival have been comparable to bone marrow transplant and the incidence of GVHD was reduced4). In the present study, 2 patients received unrelated UCBT, but both of them rejected initial grafts. Thus, caution should be taken to perform UCBT, especially if the cell number of the UCB unit is marginal or if the patient had received lots of transfusions (case 9).
There are no nation-wide data on FA incidence in Korea, but a recent retrospective study from 9 centers of Korea reported preliminary data on 41 FA patients who underwent SCT between 1996 and 200931). Thirty patients (73.2%) were alive with a median of 35 months after transplantation. Flu-based regimens were used in 28 patients (68.3%), and radiation in 21 patients (51.2%). Event-free survival differed by donor types with the best outcome of HLA-matched unrelated donor (79%, n=20), followed by matched sibling donor (67%, n=7), haploidentical parental donor (50%, n=2), and UCB (25%, n=12). The incidence of acute grades II-IV and chronic GVHD was 39% (n=16) and 19% (n=7), respectively.
Many patients who develop BMF initially respond to supportive care such as blood transfusions, androgens, and cytokines (e.g., G-CSF and GM-CSF)4). Synthetic androgens, such as oxymetholon and danazol, are often considered to be effective for treating BMF in FA patients, although long-term androgen use has side effects that include masculinization, acne, hyperactivity and increased liver tumor incidence32). In this study, a female patient (case 3) treated with androgen suffered from masculinization and one (case 10) developed peliosis hepatis. Three out of 8 patients treated with androgen showed partial responses. However, all 4 patients who received androgen therapy alone because of the lack of a suitable donor or before the availability of SCT at the institution eventually died. Among 4 patients who finally underwent a SCT, 3 are alive with a median follow up of 173.5 months.
Growth and endocrine abnormalities are clinically important aspects of FA as well. They are often either secondary to hormonal deficiencies, including pituitary hypofunction with hypogonadism, growth hormone deficiency, hypothyroidism and abnormal glucose or insulin metabolism, or treatment-related, such as hemochromatosis with repeated transfusions, or transplant-related complications3). A thorough baseline and annual endocrine evaluation should be performed in every FA patient to reduce morbidity and improve quality of life.
Despite the small sample sizes and without complementation grouping, the current study provides information on clinical manifestation and long-term outcome of Korean FA patients from a single institution. A nation-wide screening and registry for FA along with genetic subtyping should be initiated (1) to understand the possible ethnic differences in the frequency of subtypes, (2) to unravel genotype-phenotype correlation, (3) to further refine techniques of HSCT, (4) to develop potential future therapies, such as improved gene therapy, or antioxidant compounds, and (5) to establish guidelines for long-term follow-up.

 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


