

Introduction
Mucopolysaccharidosis IVA (MPS IVA; Morquio A syndrome) is an autosomal recessive lysosomal storage disease caused by a deficiency in N-acetylgalactosamine-6-sulfatase (GALNS)1,2). GALNS is one of several sulfatases required to degrade the glycosaminoglycan (GAG), keratan sulfate (KS) and chondroitin-6-sulfate (C6S). It is encoded by the GALNS gene3). In the absence of this enzyme, the stepwise degradation of KS and C6S is blocked, resulting in the intracellular accumulation of the respective GAG in the lysosomes of a wide range of tissue4). It is manifests mainly as short stature and skeletal dysplasia5) with bone deformity being the most common initial symptom6). Additional compromised systems include the visual, auditory, digestive, cardiovascular, and respiratory system7).
MPS IVA is a rare disorder, and the incidence of MPS IVA varies among different populations; reported rates range from 1 in 76,000 live births in Northern Ireland8) to 1 in 640,000 live births in western Australia9). In Japan, the incidence was 1 per 625,000 live births for five years4).
Like other lysosomal storage disorders, phenotypes of MPS IVA patients vary from the classical form with severe bone dysplasia, short trunk dwarfism, hearing loss, heart valve involvement, corneal opacity, and a life span of 20 to 30 years, to the mild form with fewer manifestations3,7). A broad spectrum of clinical severity in MPS IVA suggests allelic heterogeneity10,11). The GALNS cDNA is 2,339 bp in length with a 1,566 bp open-reading frame encoding 522 amino acids12), and the GALNS gene is split into 14 exons spanning approximately 50 kb13). To date, about 150 different mutations have been identified (Human Gene Mutation Database, www.hgmd.org).
Currently, no effective therapies exist for MPS IVA patients with severe bone dysplasia. Supportive care is used to treat the clinical manifestations of this disorder. Hematopoietic stem cell transplantation has been attempted in some patients, and enzyme replacement therapy for MPS IVA with recombinant human GALNS is now under way.
There have only been a few MPS IVA case reports conducted in Korean14), and there is a need for more research on clinical and genetic findings. In particular, the radiologic findings of Korean MPS IVA have rarely been studied even though the predominant clinical features of MPS IVA are skeletal problems. The purpose of this study was to better understand the characteristics of Korean patients with MPS IVA by studying the clinical, laboratory and particularly radiological findings of 10 patients diagnosed with MPS IVA at the Samsung Medical Center.
Materials and methods
Ten patients with MPS IVA were diagnosed between July 2004 and December 2010 at the Samsung Medical Center. Patients' charts were retrospectively reviewed for clinical features and previous medical history. Findings on physical examinations were obtained from the physicians' notes on admission or the outpatient department. Patients' radiological skeletal surveys, including skull (anteroposterior [AP] and lateral), chest (AP), spine (AP and lateral), pelvis (AP), tubular bones (AP), and hands and feet (AP), were evaluated. All radiologic findings were reading from one radiologist. Echocardiography, pulmonary function tests, and speech audiometry were also performed. For screening of corneal abnormalities, slit lamp examinations were performed by ophthalmologists.
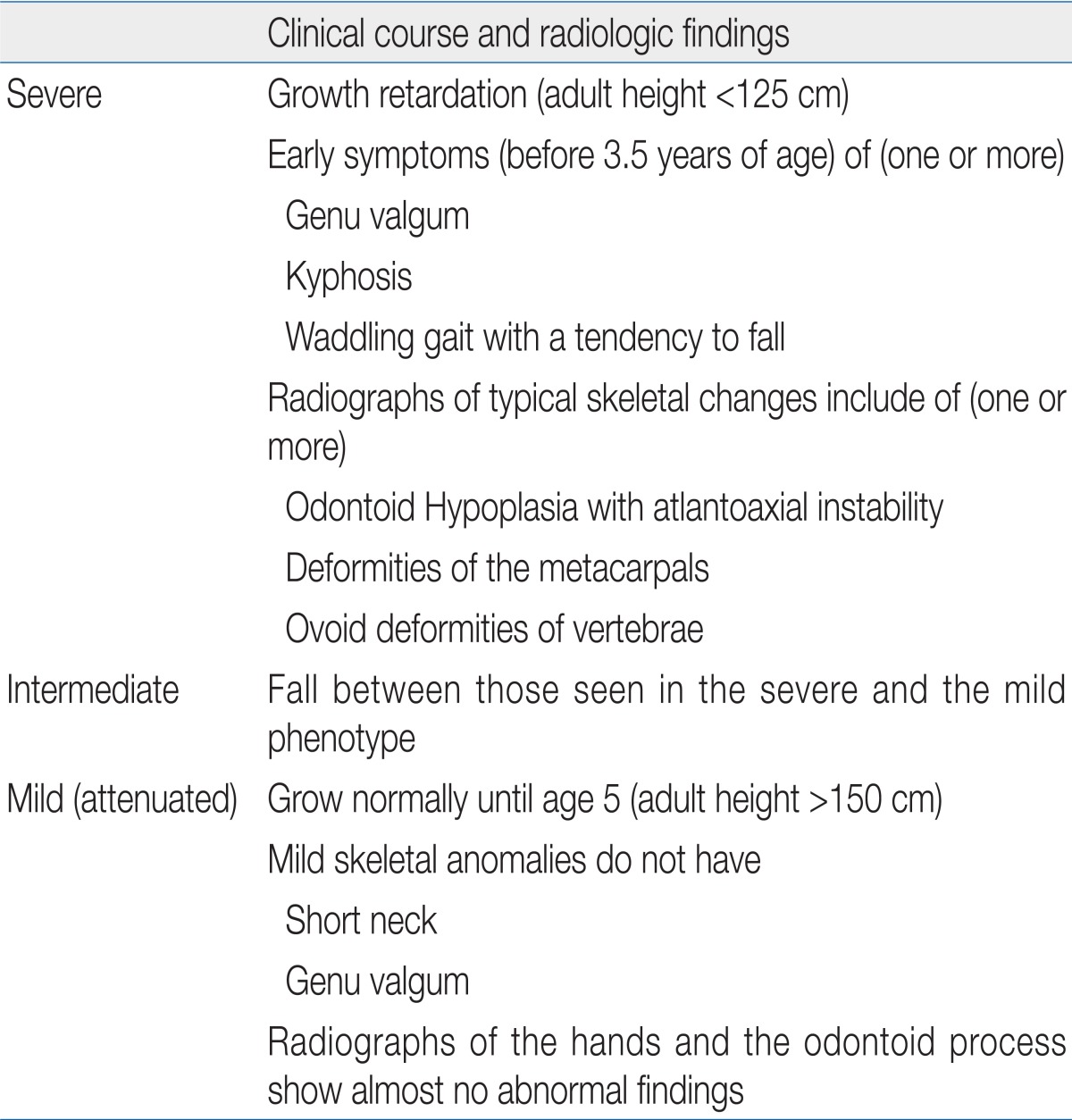
Based on previous research15,16), we classified our patients into three phenotypes by clinical manifestations determined from the physical examination, radiology, and clinical course. The criteria for each clinical phenotype is shown in Table 1.
1. Biochemical analysis
We measured urinary GAG levels using the cerylpyridinium chloride (CPC) precipitation test. To confirm MPS IVA, we performed thin layer chromatography in urine and determined the N-acetylgalactosamine-6-sulphatase activity in either peripheral blood leukocytes or skin fibroblasts. N-acetylgalactosamine-6-sulphatase activity was measured using the fluorophotometry method with artificial 4MU-beta-D-Galactopyranoside-6-sulphate substrate.
2. Mutation analysis of the GALNS gene
Genomic DNA was isolated from peripheral blood leukocytes. The 14 exons of the GALNS gene, along with their flanking intronic regions, were amplified by polymerase chain reaction (PCR). Five microliters of the amplification product were treated with 10 U shrimp alkaline phosphatase and 2 U exonuclease I (USB Co., Cleveland, OH, USA). Direct sequencing of the DNA was performed using the ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) with the BigDye Terminator Cycle Sequencing-Ready Reaction Kit (Applied Biosystems). The cDNA nucleotide sequences of the GALNS gene examined were numbered according to their respective GenBank accession numbers of NM_000512.4 for GALNS. Our mutation nomenclature follows the recommendations of the Human Genome Variation Society (http://www.hgvs.org/mutnomen/), with nucleotide +1 corresponding to the A of the ATG translation initiation codon. All novel mutations were confirmed via sequencing of 100 control chromosomes. To avoid misinterpretation of the reading frames, we used both forward and reverse sequencing. We also investigated the mutational status of the patients' family members by PCR and sequencing.
Results
1. Clinical findings
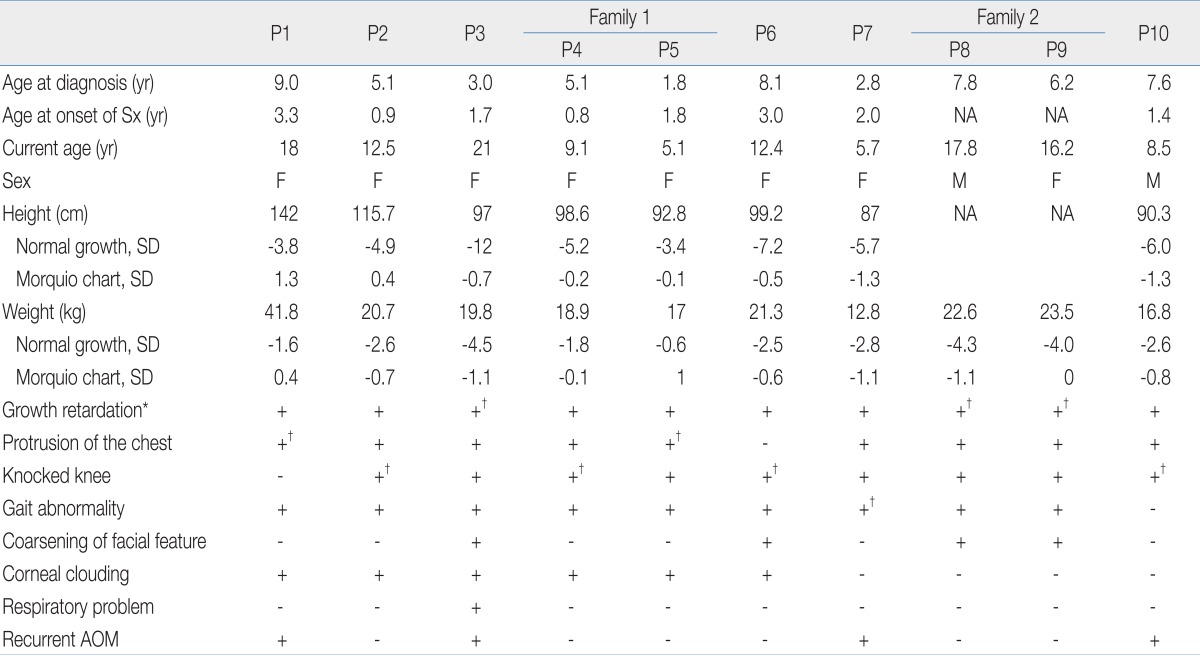
Ten patients (eight males, two females) from eight families were involved in the study. The clinical findings are shown in Table 2. The median age at onset of disease-related initial symptoms was one year and seven months (ranging from nine months to three years and three months) and median age at diagnosis was five years and seven months (ranging from one year and nine months to nine years).
All patients had uneventful birth histories and appeared normal at birth. The common disease-related initial symptoms were bony deformities (knee, spine, and ribs), short stature, and gait disturbance. At the time of clinical diagnosis, nine patients had genu valgum, and four patients (P2, P4, P6, and P10) had undergone a hemiepiphysiodesis operation. All patients had hip joint dysplasia when diagnosed and two patients (P1 and P4) had undergone hip capsulotomy and capsular placation operations. The consequence of these skeletal changes in most patients was restricted mobility of the large joints (especially the hips, knees, and elbows). Four patients (P1, P3, P8, and P9) were unable to walk, and five patients (P2, P4, P5, P6, and P7) had a waddling gait and a tendency to fall. But, No one had undergone a cervical spine operation.
All patients had growth retardation. At their current age, the heights of eight patients ranged from -12 to -3.4 standard deviation (SD) when compared to the height of age and gender-matched healthy children17). Compared with the Morquio growth chart, the heights of our patients ranged from -1.1 to 1 SD18). Two patients (P8 and P9) were unable to check their height because they could not stand. Two patients (P1 and P8) had previously undergone a ventilation tube insertion operation because of recurrent otitis media. Patient 3 received at-home ventilation care due to restrictive lung disease. Based on our criteria, nine patients were classified as having the severe form and one patient was classified as having the intermediate form of the disease.
2. Radiologic findings
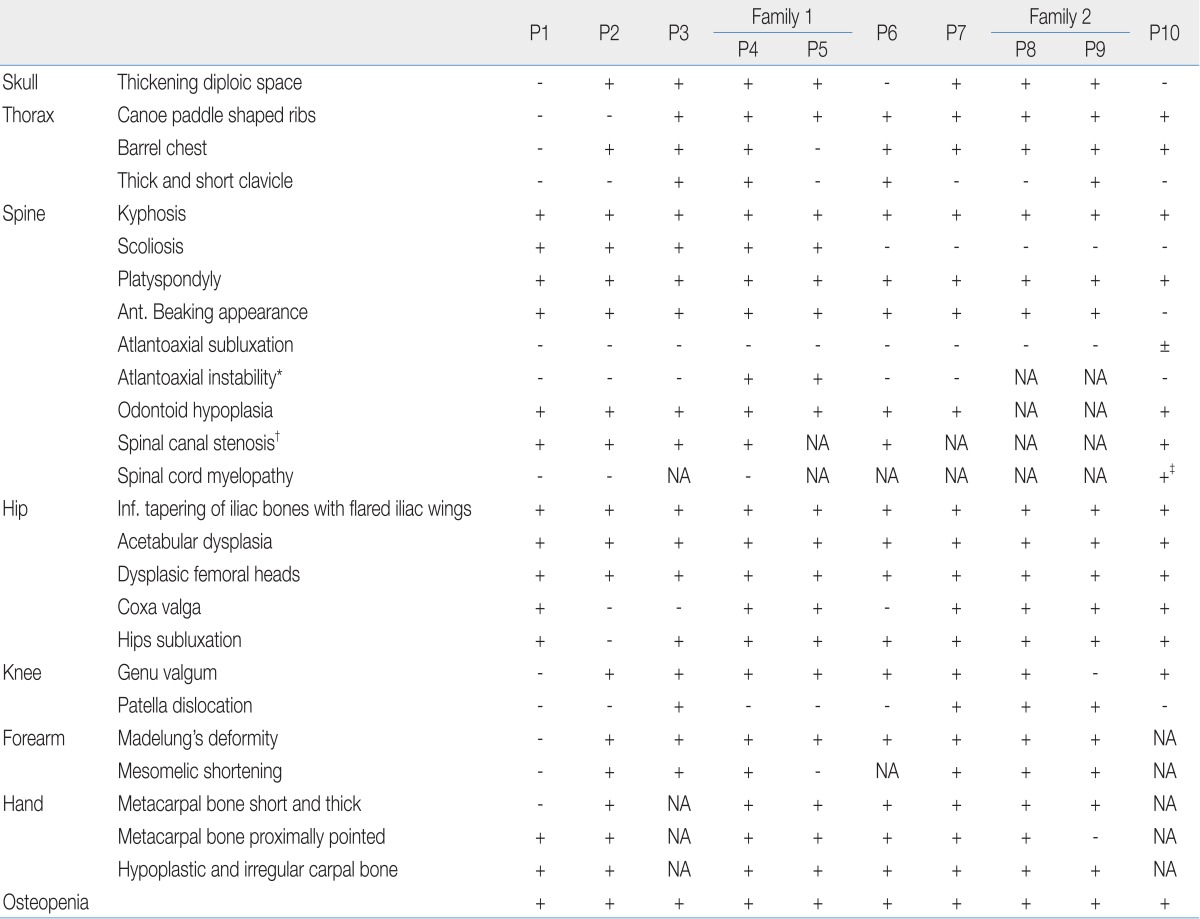
Skeletal abnormalities were present in all patients. The radiographic results are shown in Table 3. In the skull, no one had abnormal J-shaped sella turcica; seven patients had thickened diploic space. Roentgenographic findings of the chest included barrel chest deformities in eight patients and paddle-shaped ribs in eight patients.
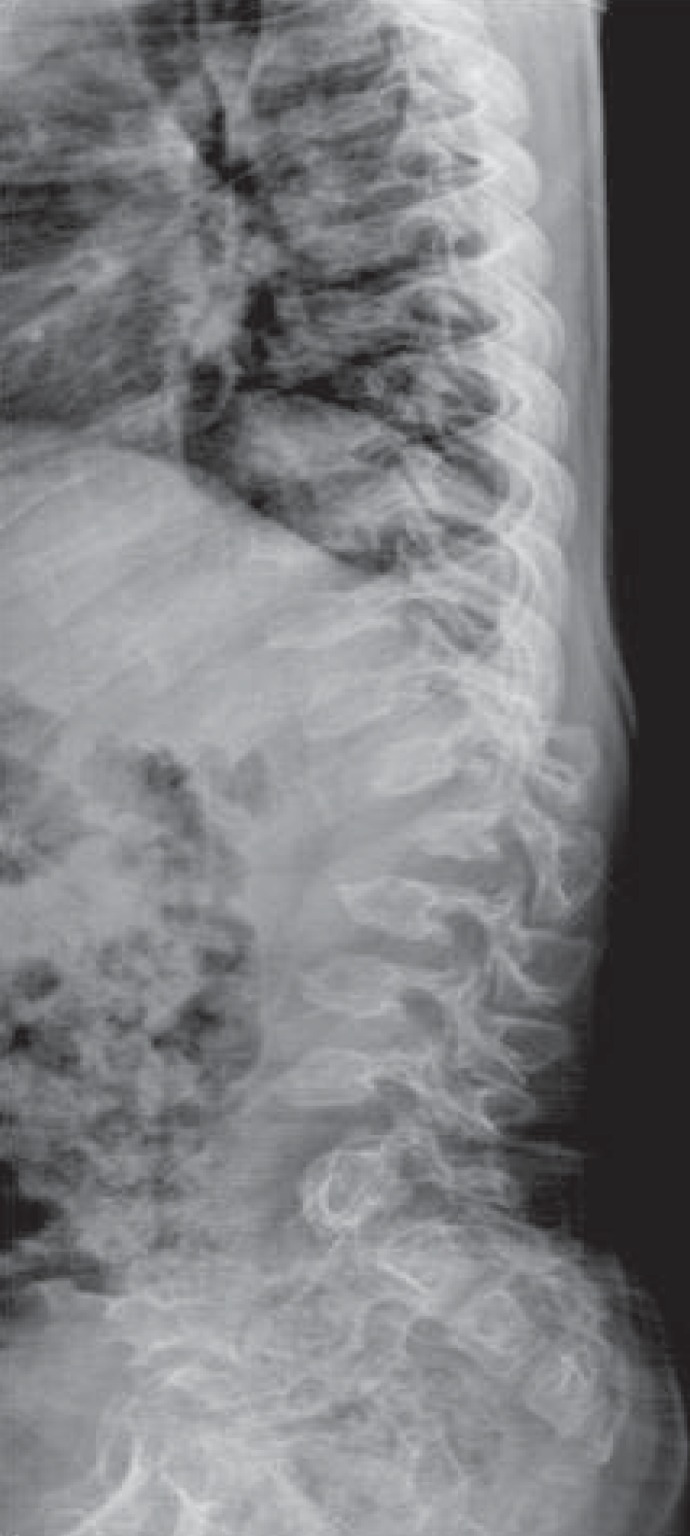
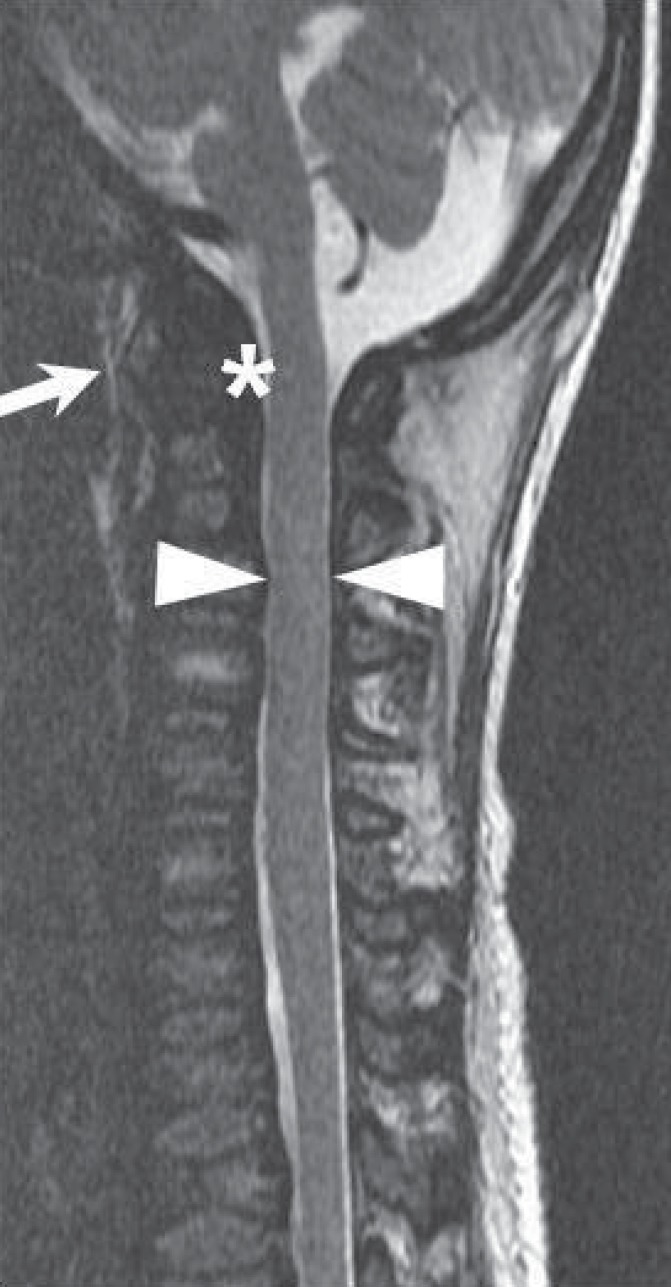
The AP radiographic view of the spine showed thoraco-lumbar scoliosis in five patients; the lateral view demonstrated thoraco-lumbar kyphosis with irregular, platyspondyly and anterior beaking vertebrae in all patients (Fig. 1). In the cervical spine, eight patients had an odontoid hypoplasia and two patients had an atlantoaxial instability that increased the predentate space more than 5 mm in the neck flexion and extension views. Patients 8 and 9 were uncooperative and did not complete the test. Six patients had spine magnetic resonance imaging (MRI) or computed tomography to gather further information and obtain an accurate evaluation of the cervical spine. All six showed spinal canal stenosis. Spinal stenosis was defined as a ratio of the spinal canal and vertebra that is below 0.8. One patient (P10) showed mild atlantoaxial subluxation and subtle signal changes in the spine MRI, which suggested cord myelopathy (Fig. 2).
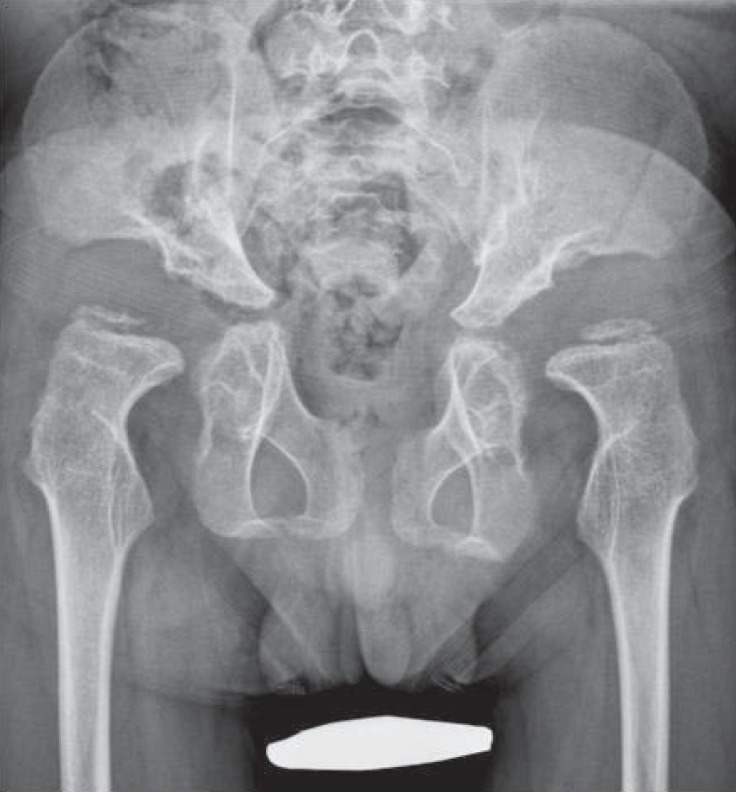
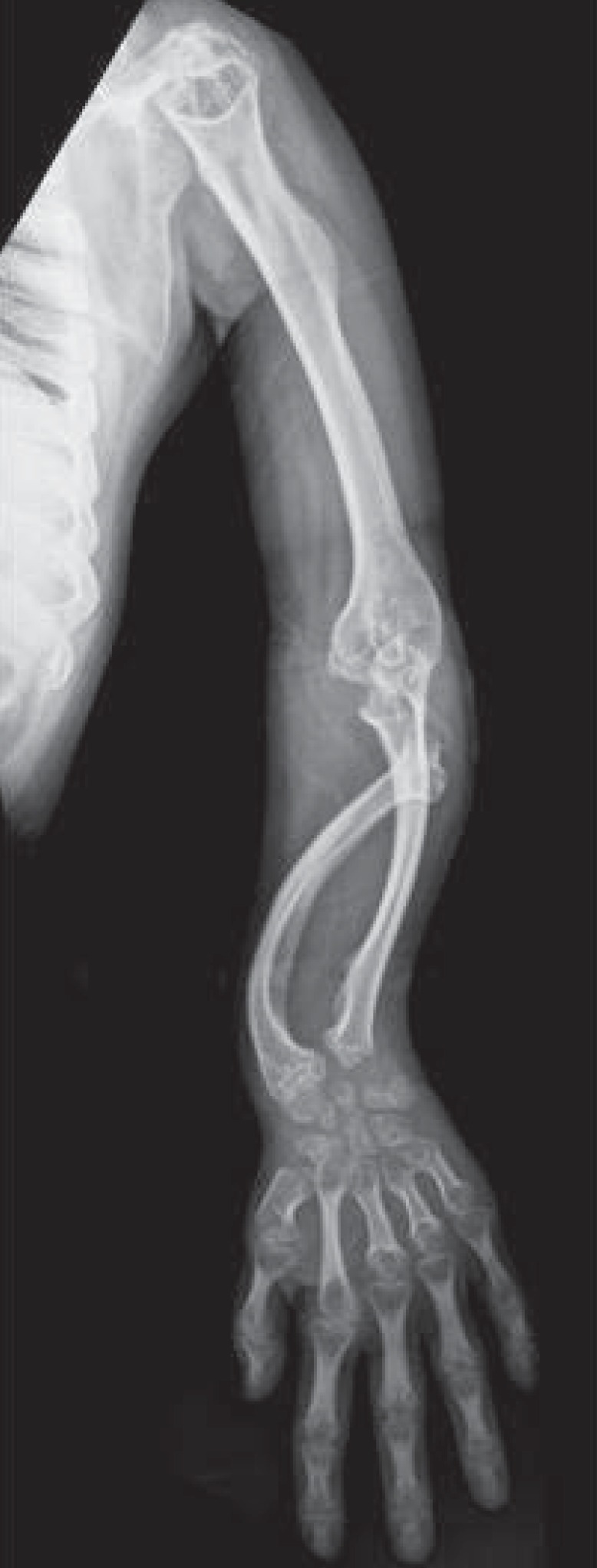
Of note, almost all of the patients' radiologic findings indicated severe bony dysplasia of the limbs. In the hip joint, all patients showed inferior tapering of the iliac bones with flared iliac wings. Acetabular and femoral head dysplasia were also observed in all patients. Even worse, coxa valga was present in seven patients and hips subluxation was observed in nine patients (Fig. 3). In the knee joint, eight patients had genu valgum and four patients had patella dislocation. In the upper extremities, eight patients had a Madelung deformity and seven patients had mesomelic shortening. In the hands, seven patients had proximally pointed metacarpal bones that appeared short and thickened. Hypoplastic and irregular carpal bone changes were observed in eight patients (Fig. 4).
In addition, all patients had mild to severe osteopenia.
3. Extraskeletal findings
Echocardiography was done in eight patients. One patient (P1) had normal echocardiogram findings, and the other patients showed mild abnormal findings like minimal mitral or tricuspid valve regurgitation or thickening of the aortic or the mitral valve. The size of the left ventricle of three patients (P4, P7, and P10) was enlarged. However, none had decreased left ventricular contractility. Pulmonary function tests were performed in five patients (P1, P2, P4, P6 and P10). All five had a normal range of forced vital capacity and forced expired volume in one second in spirometry test, and a normal flow volume curve. Because one patient (P3) was using an at-home ventilator, the test was not performed. Others could not exam due to noncooperation.
Ophthalmologic testing was performed on six patients by slit lamp examination. Five patients (P1, P2, P4, P5, and P6) had mild corneal opacity and one patient (P3) had severe opacity. Of the four patients who did not take this ophthalmologic test, corneal clouding was not observed in their general physical examinations. Four patients (P2, P6, and P10) received a speech audiometry test. One patient (P3) had a sensory neural hearing loss. Other patients couldn't test due to noncooperation. No patients had significant cognitive problems.
4. Biochemical and genetic analysis
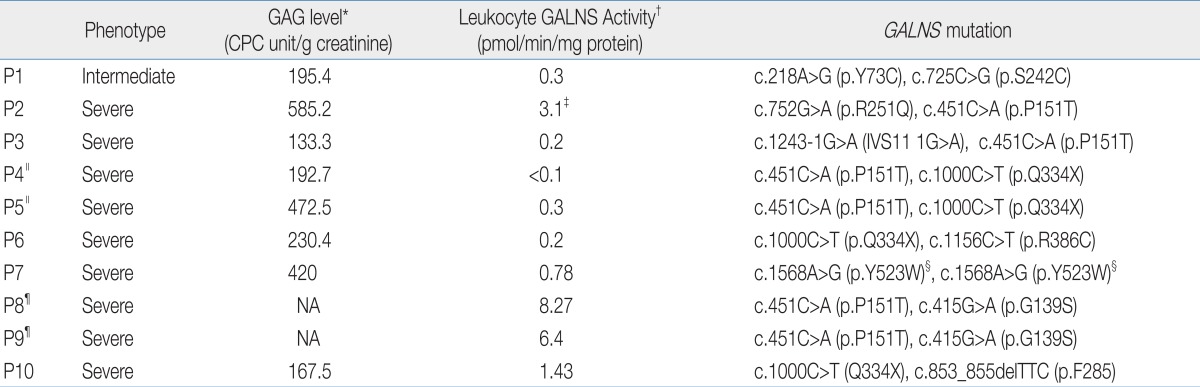
The biochemical and genetic data are shown in Table 4. The urine KS concentration was elevated in all MPS IVA patients ranging from 133.3 CPC unit/g creatinine to 585 CP C unit/g creatinine compared with normal control values. Normal ranges of GAG levels are <175 CPC unit/g creatinine (age, 1 to 9 years,) and <85 CPC unit/g creatinine (age, >9 years,) in random urine.
Nine patients showed severely reduced GALNS activity ranging from not detected to 8.27 pmol/min/mg protein in leukocytes (normal range, 39 to 166 pmol/min/mg protein), and the remaining patient exhibited 3.1 pmol/min/mg protein of enzyme activity in skin fibroblasts (normal range, 18 to 72 pmol/min/mg protein).
From the GALNS gene analysis, total of 16 mutant alleles from 8 families were identified, corresponding to 10 different mutations (Table 4); seven missense mutations of c.218A>G (6.3%, 1/16), c.415G>A (12.5%, 2/16), c.451C>A (37.5%, 6/16), c.725C>G (6.3%, 1/16), c.752G>A (6.3%, 1/16), c.1156C>T (6.3%, 1/16), c.1568A>G (12.5%, 2/16), one nonsense mutation of c.1000C>T (25%, 4/16), one deletion mutation of c.853_855delTTC (6.3%, 1/16), and one splice site mutation of c.1243-1G>A (6.3%, 1/16). The c.725C>G and c.752G>A mutations were in exon 7, and the other mutations were scattered in different exons or introns. The c.451C>A and c.1000C>T account for 37.5% (6/16) and 25% (4/16) of the total number of mutant alleles in the Korean patients with MPS IVA, respectively.
One mutation of c.1568A>G (p.Y523W) in patient 6 was a novel homozygous mutation. But we didn't have approval of family study in this patient.
Discussion
In our study, nine patients were classified as having the severe phenotype and one patient was classified as intermediate. All patients had growth retardation and their height at current age ranged from -12 to -3.4 SD when compared to the height of age and gender-matched healthy children. And all patients showed severe skeletal dysplasia on radiologic findings, especially in their hips and limbs. Consequently, some patients had flexion deformities of the arms, were unable to walk, and underwent knee or hip joint operations. However, MPS IVA patients are usually not evaluated for unusual skeletal features until their second or third year of life. Thus, the average diagnostic age differs significantly from the age of onset6).
Compared with other patients who have MPS, patients with MPS IVA tend to have greater spine involvement with scoliosis, kiphosis, and severe gibbus19). Odontoid hypoplasia is the most critical skeletal feature in any patient with Morquio syndrome. In fact, both mortality and morbidity are related primarily to atlantoaxial subluxation resulting from the instability of the odontoid process19). Therefore, several studies have suggested that patients who have an increased risk of developing symptoms of spinal cord compression require the application of a brace or an early prophylactic posterior cervical fusion20,21). One MPS IVA patient, who was diagnosed at Samsung Medical Center but not included in this study, expired due to atlantoaxial subluxation, which occurred while she was washing her hair. All patients included in our study, except two patients who were unable to be fully examined, had odontoid hypoplasia, and two patients had atlantoaxial instability. In addition, one patient showed mild atlantoaxial subluxation and cord compression in a spine MRI, but he had no neurologic symptoms like cord paralysis or sphincter dysfunction. In our study, the cervical instability and subluxation were less common than previous study4) reported that 90% of the MPS IVA patients had cervical instability and cervical spine operations were the most frequent surgical procedure in MPS IVA patients.
The genetic analysis, 10 different GALNS mutations were identified. The p.R386C mutation was the most common mutation worldwide, and it has been found in 34 alleles from 25 patients of American (Caucasian), Argentine, Brazilian, British, Chilean, Colombian, German, Italian, Japanese, Mexican, Polish, and Turkish origin22). In our study, the p.R386C mutation accounted for only 6.3% (1/16) of all mutations. However, p.P151T and p.Q334X mutations accounted for 37.5% (6/16) and 25% (4/16) of all mutations in this sample, respectively. These two mutations have been found in Korean patients (Pack et al., unpublished data).
A limitation of this study was the small number of patients involved, which can be explained by the fact that MPS IVA is an extremely rare disease and accounts for about 4 percent of all Korean patients with MPS23). Additionally, we were unable to determine the correlation between phenotype and biochemical data because nine patients had a severe phenotype, only one patient was intermittent, and no patients showed a mild phenotype.
As shown above, the Korean patients with MPS IVA had severe skeletal problems and almost all had a severe phenotype by the time they were diagnosed. This means that those patients with mild symptoms or deformities did not suspect MPS, thus did not take the MPS screening test. With a more accurate understanding of clinical and radiological features of MPS IVA, however, we can identify the symptoms earlier. This will lead to more patients being diagnosed earlier, so proper therapy can be provided and irreversible damage can be prevented. Furthermore, as with other types of MPS, research on enzyme replacement therapy in MPS IVA is currently in progress24,25).
Patients with MPS IVA can be distinguished from patients with other MPSs because their intelligence is preserved, but they have severe skeletal abnormalities24). Therefore, adequate evaluations and therapy in the early stages of the disease can greatly improve the quality of life of patients suffering from skeletal abnormalities. Moreover, such early detection and intervention can reduce the mortality rate of those with atlantoaxial subluxation.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


