

Introduction
Takayasu arteritis (TA) is a rare disease of unknown etiology, characterized by chronic inflammation of large and medium-sized arteries, particularly the aorta and its major branches1). TA usually occurs in women in their second or third decade of life and more commonly in Southeast Asia, but now it has been recognized worldwide in both sexes2). Vascular inflammation may cause arterial stenosis, occlusion, thrombosis, dilatation, or aneurysms and may further induce clinically varied symptoms according to sites of involved vessels3). Aortic regurgitation (AR) is a potentially fatal complication of TA and may result from involvement of the ascending aorta or the aortic valve itself3). Here, we report a very rare case of infantile TA and severe aortic insufficiency, which required aortic valve replacement surgery in a young boy.
Case Report
A 10-month-old boy was admitted to another hospital due to excessive sweating and lethargy. Echocardiography showed cardiomegaly and decreased left ventricular (LV) function. The child was initially suspected of having myocarditis because he had exhibited a mild cough, rhinorrhea, and diarrhea 1 month prior to admission. He was treated with intravenous inotropic agents and other medications for heart failure, including enalapril, carvedilol, furosemide, and spironolactone. Within 12 days, his clinical symptoms were brought under control, and he was discharged from the hospital.
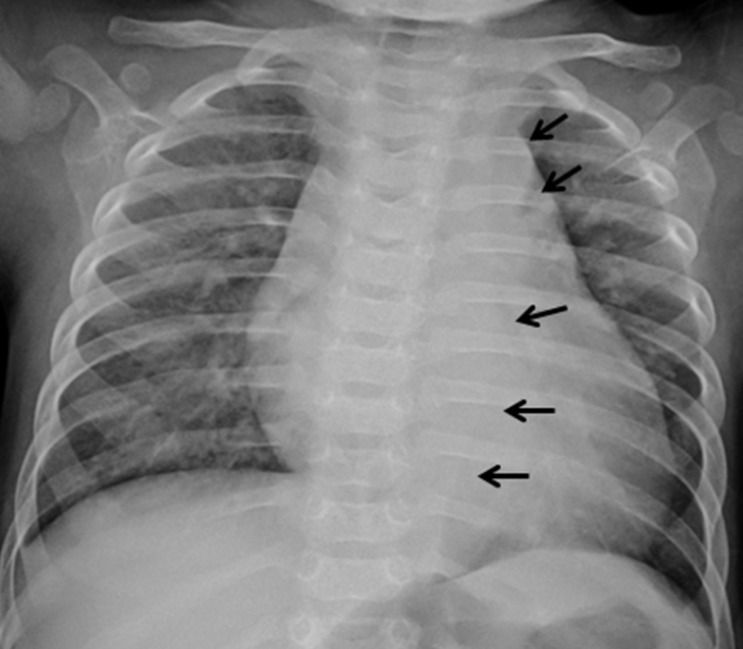
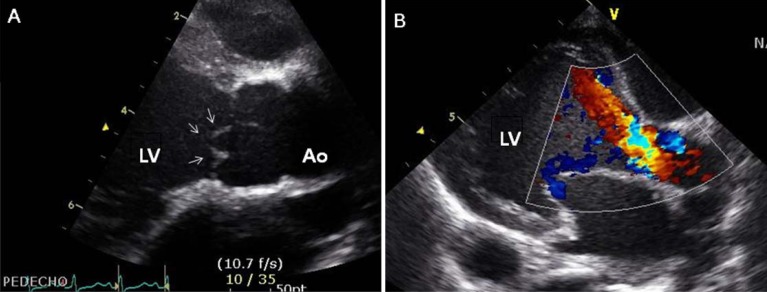
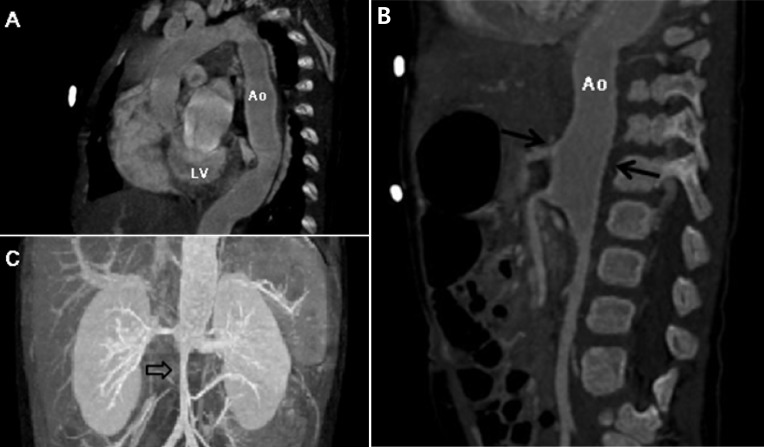
Two days after his discharge from the first hospital, he was brought to Seoul National University Children's Hospital exhibiting anorexia and lethargy. At the time of admission, he was 76.4 cm tall and weighed 9 kg. Physical examination revealed a grade 3/6 diastolic murmur audible at the left lower sternal border. Peripheral pulses were bounding and symmetric. Blood pressure was 103/38, 101/46, 100/42, and 109/40 mmHg in the right arm, left arm, right leg, and left leg, respectively. A chest radiograph revealed cardiomegaly and an irregular contour of the lateral margin of the descending thoracic aorta (Fig. 1). His first echocardiography at our institution showed severe AR with a dilated LV end-diastolic dimension (LVEDd, 45.5 mm) and borderline LV systolic function (ejection fraction [EF], 58%). Dilatation was observed in the aortic valve annulus (15.8×17.7 mm), aortic sinuses (maximum diameter, 20.3 mm), sinotubular junction (16.5 mm), and ascending aorta (17 mm). Aortic leaflets were prolapsed, lacking coaptation during diastole and hence producing multiple, severe leaks (Fig. 2). Results of the echocardiography also revealed a markedly dilated thoracoabdominal aorta. Computed tomography (CT) angiography (Fig. 3) was performed to evaluate the aortic arch and its major branches; CT showed an abnormally dilated thoracic and abdominal aorta (largest diameter, 17.8 mm) with diffuse narrowing of the infrarenal aorta (diameter, 3.4 mm), although no significant stenosis was observed in either renal artery. Whole-body 18F-fluorodeoxyglucose positron emission tomography (18F-FDG-PET), performed to identify disease activity related to aortitis, indicated moderate diffuse hypermetabolism in the aorta from the aortic arch to the abdominal aorta adjacent to the kidneys. These findings were consistent with arteritis involving large arteries. The patient also had a leukocyte count of 4,250/µL; hemoglobin level, 10.2 g/dL; erythrocyte sedimentation rate (ESR), 89 mm/hr; and C-reactive protein, 0.38 mg/dL. Cardiac enzyme levels were elevated; creatine kinase, myocardial bound was 7.0 ng/mL and troponin-I was 1.03 ng/mL. The brain natriuretic peptide level was 2,888 pg/mL. Serologic tests for syphilis, coxsackie virus, adenovirus, echovirus, and Epstein-Barr virus were negative. Polymerase chain reaction for enterovirus, influenza virus, and respiratory syncytial virus were also negative. Serum hepatitis B surface antigen, anti-nuclear antibody, and anti-double-stranded DNA were nonreactive. A clinical diagnosis of TA was established based on the combination of aortic dilatation, hypermetabolism of the aorta, and elevated ESR. The patient was initially treated with prednisolone (60 mg/m2) from the seventh day following his admission. One week after initiation of this treatment, administration of prednisolone was reduced to 1 mg·kg-1·day-1 (approximately 20 mg/m2) for 2 additional weeks. During this period, we observed a reduction in inflammatory activity of the disease, as ESR decreased from 89 mm/hr to 11 mm/hr. However, serial echocardiography over the subsequent 1-month interval indicated deterioration of the patient's cardiac function, as his LVEDd increased from 45.5 to 53.2 mm and LV EF decreased from 58 to 53.7%.
The patient then underwent surgical commissuroplasty for the severely prolapsed aortic valve on hospital day 34. During surgical exploration, slight dilation of the ascending aorta was observed, but thickening or inflammation of the aortic wall was not observed. However, pathological examination of an aortic wall specimen did reveal intimal thickening related to myxoid degeneration with a few lymphocytic infiltration. The patient's AR improved from grade 4/4 to 2/4 immediately after the commissuroplasty; however, within 1 month of the surgery, AR was aggravated again, and both LV dilatation and systolic dysfunction were observed. Eventually, the patient underwent aortic valve replacement surgery and received a 17-mm St. Jude mechanical aortic valve on hospital day 68. An aortic valve specimen from the surgery showed myxoid valvular thickening without vascular proliferation. The patient was discharged 13 days following aortic valve regurgitation, and his condition has stabilized due in part to continuing post-surgery treatment with enalapril, losartan, carvedilol, digoxin, furosemide, spironolactone, prednisolone (0.6 mg/kg per day), and warfarin (0.5 mg/kg per day).
Discussion
This case is interesting because of the early and unusual onset of TA in infancy. The association between severe aortic valve regurgitation and its management is also worth reporting. The incidence of TA-associated AR is not uncommon and has been reported in 18 to 55% of all TA cases1,4-7), with the youngest case ever reported that of a 2-year-old boy8). AR can be attributed to both aortic dilation with separation of aortic commissures and direct inflammation of the aortic leaflets4,6). AR can induce congestive heart failure or arrhythmia, both of which are the main causes of death in patients with TA9). Therefore, active follow-up and aggressive treatment is important for severe AR.
Diagnosis of TA is often delayed because clinical presentation of TA varies depending on the location of vessel involvement. The median delay between onset of symptoms and diagnosis is from 10 months to 4.9 years1,5,10,11). Various clinical manifestations of TA include hypertension; systemic symptoms, such as malaise, fever, night sweating, and anorexia; and ischemic complications in the territory of occluded vessels, such as abdominal pain after eating, headache, impaired vision, or limb claudication12). This patient showed only nonspecific symptoms of heart failure, and he was too young to complain of associated ischemic symptoms; this impeded diagnosis.
Early diagnosis of TA is important because early treatment may prevent progression of vascular lesions. However, no single laboratory test is available to confirm the diagnosis of TA. Diagnosis is determined based on the combination of history, physical findings, clinical suspicion, and vascular imaging techniques1,13). According to recently proposed criteria, diagnosis of childhood TA requires identification of typical angiographic abnormalities of the aorta or its main branches (mandatory criterion) plus 1 of 5 additional criteria: 1) pulse deficit or claudication, 2) blood pressure discrepancy in any limb, 3) bruits, 4) hypertension, and 5) elevated acute phase reactant14). Conventional angiography is still considered as the gold standard for diagnosing TA; however, because conventional angiography is an invasive procedure with high exposure to radiation, this modality is not frequently used in clinical settings15). We utilized CT angiography for evaluating the aorta and its main branches and introduced a new imaging technique, 18F-FDG-PET, which has been recently used in the diagnosis of TA16,17). 18F-FDG-PET is noninvasive and can be used to detect early disease manifestation by identifying metabolic changes in the vessel wall15).
Although definite diagnosis of TA is possible when the pathology of the involved vessel shows characteristic features of TA, histologic assessment is limited to patients undergoing surgery12). In the acute phase of TA, vascular media is infiltrated with lymphocytes and occasional giant cells with neovascularization. Additionally, vascular intima is thickened with mucopolysaccharides, smooth muscle cells, and fibroblasts. In the chronic phase of TA, fibrosis with destruction of elastic tissue can be observed12). In this case, pathological examination of an aortic wall specimen showed intimal thickening related to myxoid degeneration with a few lymphocytic infiltration, but there was no other evidence of active inflammation. Absence of acute inflammatory change in the patient's aortic wall specimens suggested that inflammation was well controlled by the preoperative steroid treatment. Nishimura et al.18) have conducted a detailed pathological examination of one case after aortic valve replacement surgery for TA complicated by AR, and the aortic specimen revealed a similar result to ours. Also, absence of abnormalities in biopsy findings did not rule out TA, if other forms of arteritis were excluded. Lupi-Herrera et al.13) have reported that a positive result of arterial biopsy was observed in only 35.7 percent of the patients (15/42) in a clinical study of 107 cases with TA.
In children with suspected TA, some more common diagnoses have to be considered. The differential diagnoses include developmental disorders (coarctation of aorta and interruption of aortic arch variant), genetic vasculopathies (Marfan syndrome and Loeys-Dietz syndrome), other autoimmune disorders (Behcet's disease, lupus, rheumatoid arthritis and giant cell arteritis), or infectious aortitis (syphilis)12). In this patient, we excluded coarctation of aorta and interruption of aortic arch variant because his aortic arch was dilated rather than stenotic. We also excluded Marfan syndrome because of diffuse narrowing of the infrarenal aorta. Loeys-Dietz syndrome, an autosomal dominant disorder of connective tissue, may also be present with an aortic aneurysm during infancy, we excluded this syndrome because the patient did not have typical craniofacial manifestations, such as hypertelorism, cleft palate, or bifid uvula19). Furthermore, such developmental disorders and genetic vasculopathies do not show abnormal uptake in the aortic wall on 18F-FDG-PET. Other autoimmune disorders have specific features that enable diagnosis, but this patient did not show any features compatible with the disorders. We ruled out lupus because serum anti-nuclear antibody and anti-double-stranded DNA were nonreactive. Syphilis was excluded by a negative result of serologic test for syphilis.
The prognosis for TA is variable and appears to depend on the rate of disease progression as well as the presence of other major complications such as AR, retinopathy, hypertension, or aneurysm formation10). Because this patient displayed rapid disease progression coupled with severe AR, early aortic valve replacement was recommended. To prevent detachment of the prosthetic valve or formation of an anastomotic aneurysm, control of inflammation would be crucial in this patient. Glucocorticoids are mainly used as anti-inflammatory therapy for active TA. However, relapse is common with tapering and withdrawal of corticosteroids20). Previous reports have demonstrated that approximately 50% of all patients exhibit glucocorticoid resistance or suffer relapse, thus necessitating treatment with additional immunosuppressive agents such as azathioprine, methotrexate, or cyclophosphamide3,11). Furthermore, long-term side effects of corticosteroids may be extensive, so the adjunctive use of immunosuppressants may allow administration of reduced doses of corticosteroids for patients with TA.
In conclusion, to our knowledge, this patient is the youngest reported case of TA-associated severe AR. TA should be considered in a child who presents with unexplained systemic symptoms coupled with a cardiac murmur. Complete evaluation of the entire aorta with CT angiography and 18FDG-PET enabled confirmation of TA. Because AR is a potentially fatal complication of TA, early and aggressive aortic valve replacement surgery with perioperative immunosuppressive therapy should be considered.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


