

Introduction
Prader-Willi syndrome (PWS) is a complex multisystem genetic disorder characterized by hypothalamic-pituitary dysfunction1,2). The main clinical features include neonatal hypotonia, distinctive facial features, delayed overall development with mental deficiency, behavioral abnormalities, and poor growth in infancy, followed by overeating with severe obesity, short stature, and hypogonadism later during development3). Both the genetic and endocrine aspects of this condition have garnered interest, with growth hormone (GH) therapy receiving particular attention in recent years. Our aim is to provide an update on the recent findings regarding the genetic aspects of PWS, a thorough analysis of endocrine dysfunction in PWS (GH deficiency, obesity, sexual development, hypothyroidism, and adrenal insufficiency), and the therapeutic effects of GH treatment.
Genetics of PWS
The various genetic changes that cause PWS also lead to the loss or failure of expression of paternally expressed genes on chromosome 15q11.2-q13; this is because the maternal contribution has been programmed by epigenetic factors (e.g., DNA methylation) to be silenced4). Three mechanisms have been described: paternal deletion, maternal uniparental disomy (mUPD), and deficient imprinting3). Parent-of-origin-specific DNA methylation can be used to confirm the clinical diagnosis of PWS patients of all 3 molecular classes. The most widely used DNA methylation test targets the 5' end of the small nuclear ribonucleoprotein polypeptide N (SNRPN) locus5,6). Normal individuals have both a methylated and an unmethylated allele, whereas individuals with PWS have only the maternally methylated allele.
1. Microdeletion of the chromosome region 15q11-q13
De novo deletion of the proximal region of the paternal chromosome 15 [del(15)(q11-q13)] is found in the majority (-70%) of patients with PWS. The deletion type is typically diagnosed using the SNRPN fluorescent in situ hybridization probe4). Testing for a deletion should include chromosome analysis, as the deletion is occasionally the result of a chromosomal translocation.
2. Uniparental disomy of chromosome 15
The second most common genetic abnormality in PWS (-25 to 30%) is an mUPD of chromosome 15. mUPD prevents the expression of imprinted genes that are active only on the paternal chromosome and is diagnosed using DNA polymorphism analysis of the proband and parental DNA. Four possible mechanisms can give rise to mUPD: trisomy rescue (TR), gamete complementation (GC), monosomy rescue (MR), and postfertilization mitotic nondisjunction (Mit). TR/GC is caused by nondisjunction at maternal meiosis 1 (M1) or 2 (M2), and M1 errors are associated with increased maternal age.
3. Imprinting defect
Disruption of the imprinting process on the paternally inherited chromosome 15 is the third molecular mechanism underlying PWS (2 to 5%). Most imprinting defects are epigenetic (epimutations) and exhibit a maternal-only DNA methylation pattern, despite the presence of both parental alleles. DNA sequence changes are not found in these epimutations. By contrast, approximately 15% of individuals with an imprinting defect are found to have a very small deletion in the PWS imprinting center (IC) region located at the 5' end of the SNRPN gene.
4. Classification of the (epi) genetic subtype in Korean PWS
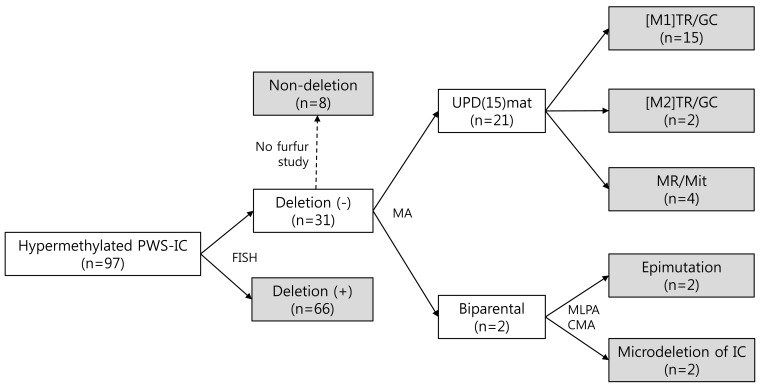
Recently, we classified a cohort of 97 Korean patients at the Samsung Medical Center, comprising 66 PWS patients with deletions and 31 PWS patients without deletions (Fig. 1). Microsatellite genotyping was performed in 23 of the 31 patients without deletions, and 15 patients were classified in the [M1]TR/GC group, 2 patients in the [M2] TR/GC group, and 4 patients in the MR/Mit group. Two patients were classified as having epimutations because microdeletions affecting the PWS IC were not identified by multiplex ligation-dependent probe amplification or chromosomal microarray. In addition, we found an increasing tendency for [M1]TR/GC caused by advanced maternal childbearing age in Korea. The maternal ages were significantly higher in the [M1]TR/GC group than in the deletion group, which was not associated with any maternal age effect. The advanced maternal ages at birth since 2006 were primarily associated with the high frequency of [M1]TR/GC. The increased number of mUPD PWS births in Korea is thought to represent an advanced economy and a trend toward having families later in life. If we assume that the number of patients with PWS caused by deletions is not decreasing, we would expect a small increase in the incidence of PWS, given the increase in the proportion of those with mUPD.
GH deficiency in PWS
More than 85% of patients with PWS are have GH deficiency7). Although height at birth is normal, growth velocity is significantly decreased after 2 to 3 years, and the mean final adult height is 2 standard deviation scores below normal8). Many features of PWS are indicative of deficient GH production, including low growth velocity despite obesity, reduced lean body mass (LBM), low insulin-like growth factor-I (IGF-I) levels, and low insulin levels7,9). These findings provide a rationale for GH therapy in PWS.
1. Effects of GH therapy
1) Height and growth velocity
With GH therapy, the overall height gain in PWS patients is similar to that seen in patients with GH deficiency and is greater than in non-GH-deficient patients of short stature, even those administered with high dosages10). GH treatment at the currently recommended dosage for children with PWS (0.03 mg┬Ękg-1┬Ęday-1) has been shown to restore linear growth and normal final adult height without inducing serious adverse effects11-13).
2) Body composition
Low LBM in PWS patients most likely reflects reduced muscle mass and may therefore contribute to clinical hypotonia, poor physical performance, and, as a result, reduced energy expenditure7). In a randomized controlled GH trial in PWS patients, LBM-corrected for height and sex- did not increase during GH treatment but significantly decreased in the control group, suggesting that GH administration prevents a reduction in LBM11). Some studies have reported major increases in bone mineral density (BMD) with GH treatment14). Other studies15,16) suggest a normal total body BMD and no effect of GH treatment on total body BMD in young children with PWS.
3) Heart
Cardiac events have been implicated in the sudden death of children and young adolescents with PWS, with or without GH treatment17,18). GH secretory status and GH therapy influence cardiac muscle mass and function19). In a retrospective study20), the response of cardiac dimensions to GH treatment was similar in PWS patients and control patients with GH deficiency, with a trend towards more elevated left ventricular parameters in PWS patients. Regular echocardiographic assessment may be considered for PWS children who undergo treatment for a prolonged duration.
2. GH therapy in infants with PWS
A randomized GH study in infants with PWS showed a significant improvement of mental/motor development in the GH group as compared with the control group21). IGF-I levels increased rapidly during GH treatment from below the normal range to the high-normal range. Theoretically, IGF-I may directly influence the central nervous system or GH may induce local IGF-I expression in brain tissue, thereby improving psychomotor development21). With improved motor development, children are able to sit, stand, and walk independently, and can explore and interact with the environment, resulting in subsequent improvement in mental development21). In addition, early GH therapy may significantly alter the natural progression of PWS by reducing body fat and improving muscle strength, physical function, and lipid profiles without resulting in adverse effects22). Early GH therapy is therefore important. At the Samsung Medical Center, nearly 200 patients have been diagnosed with PWS, and approximately 100 of these patients were diagnosed during infancy. Although children younger than 2 years are not covered by health insurance in Korea, GH therapy has been initiated in recent years in infants after obtaining informed consent from their parents. Their growth rate (in terms of height) and development (relative to chronologic age), have improved with GH therapy. Subjectively, the GH-treated infants and toddlers were reported by their families to be more alert and energetic. No serious side effects have been reported. Consequently, we recommend that GH treatment be initiated at 4 to 6 months of age with a low dose of 0.009 to 0.012 mg/kg per day, with subsequent increments during the first weeks and months to arrive at the standard replacement GH dose of approximately 0.035 mg/kg per day.
3. Concerns regarding GH therapy
1) Carbohydrate metabolism
Fasting insulin levels have been found to be lower in children with PWS than in body mass index (BMI)-matched children with simple obesity10). These low insulin levels suggest increased insulin sensitivity, probably related to GH deficiency7). Adiponectin is an adipokine that is primarily secreted from adipocytes; it has been shown to have beneficial effects in the prevention of the development of inflammatory and atherosclerotic disease23-25). The effects of adiponectin are mediated by adiponectin receptors (ADIPORs), namely ADIPOR1 and ADIPOR2. Increased expression of ADIPOR2 was found in children with PWS5), whereas ADIPORs expression in lymphocytes was markedly reduced in obese patients26). These results may account for the increased insulin sensitivity in patients with PWS, in contrast to insulin resistance observed in patients with simple obesity.
In PWS patients, the levels of total ghrelin and acylated ghrelin were increased, and these hormones are regulated by insulin. In a study on PWS children, acylated ghrelin was sensitively suppressed by insulin during an oral glucose tolerance test; this suppression was also correlated with insulin sensitivity27). In contrast, plasma obestatin, a peptide hormone derived from the proteolytic cleavage of ghrelin preprohormone28), was not elevated in PWS children and was not regulated by insulin in either PWS children or obese controls28). Insulin resistance in children with PWS may be associated with BMI even in the absence of GH treatment29). In a prospective cohort study of PWS children29), despite the favorable effect of GH on body composition and lipid profile, insulin increased during therapy. Therefore, carbohydrate metabolism (glucose and hemoglobin A1c) should be closely monitored in patients receiving GH treatment.
2) Scoliosis
Scoliosis, attributed to a combination of obesity and muscular hypotonia, is common in both children and adolescents with PWS. The prevalence of scoliosis in PWS is high (30% before age 10, 80% after age 10)30-32). The rapid growth associated with GH may aggravate this spinal deformity. Some authors have described an association between increased GH levels and a higher rate of curve progression in children without PWS33-35). In contrast to these reports, a randomized controlled trial in a large group of children with PWS showed no significant difference between GH-treated children and randomized controls with regard to the onset of scoliosis, curve progression, and start of treatment of scoliosis36). Frequent physical examinations and yearly radiographic examinations are recommended, regardless of GH treatment.
3) Obstructive sleep apnea (OSA)
Patients with PWS are also at risk of developing OSA for several reasons, including obesity37), facial dysmorphism (including micrognathia and a small naso- or oropharynx)38), sticky secretions39), and hypotonia40). Patients with PWS are reported to have elevated levels of adiponectin5), an adipose tissue-derived hormone, which has shown a negative association with OSA syndrome6). One hypothesis proposes an association between GH therapy and death during upper respiratory infections, in which GH treatment may cause enlargement of the tonsils and adenoids, which would narrow the already small airway of children with PWS41). Another hypothesis maintains that fluid retention early in therapy is the mechanism underlying the development of OSA symptoms42). A pragmatic approach in which the balance of benefit versus risk favors treatment with GH is to closely monitor each individual for OSA symptoms and to repeat the polysomnogram as clinically indicated43).
4. GH receptor polymorphism and height in children with PWS
A common GH receptor (GHR) polymorphism, reflecting the presence (fl) or absence (d3) of exon 3, has been under intensive investigation to determine its influence on response to GH therapy. Dos Santos et al.44) showed that the d3 polymorphism was associated with increased responsiveness to GH. A recent study assessed the association of GHR alleles with height before beginning GH therapy in patients with PWS45). Even before GH therapy, the d3 allele appeared to increase the responsiveness of GHRs to endogenous GH already present in patients with PWS; this response may explain why subjects with the d3 allele were taller than those who did not have the allele.
Obesity
1. Nutritional phase
PWS has been classically described as having 2 nutritional stages: failure to thrive in infancy, followed by hyperphagia in later childhood. One study described a total of 7 nutritional phases of PWS: "Decreased fetal movements and lower birth weight", "Hypotonia with difficulty feeding (0 to 9 months)", "No difficulty feeding and appropriate growth according to growth curves (9 to 25 months)", "Weight increasing without an increase in appetite or excessive calories (2.1 to 4.5 years)", "Weight increasing with an increase in appetite (4.5 to 8 years)", "Hyperphagic, rarely feels full (8 years adulthood)", and "Appetite is no longer insatiable (adulthood)"46).
2. Mechanism of hyperphagia
Several theoretical models attempt to explain overeating by PWS patients: it is a problem of satiety as opposed to hunger, it is a consequence of inner physiological awareness that has potential effects on feelings of hunger and satiety, it represents a hyperresponsive reward syste m with food as a substance of abuse, it is a direct consequence of genetics on the hypothalamic feeding pathway, and it is a consequence of the prenatal environment47). One study analyzed 18F-fluorodeoxyglucose positron emission tomography images of PWS children and found metabolic abnormalities in brain regions that are directly or indirectly related to food intake and obsessive-compulsive behavior48). In recent years, understanding hormonal mechanisms that control appetite and eating in PWS patients has generated a lot of interest.
Ghrelin, which stimulates hunger, is principally synthesized in the stomach. The number of ghrelin-expressing cells and the amount of ghrelin in the gastric body and fundus of PWS patients was 2- to 3-fold higher than that in control groups49). However, gastric emptying in PWS patients was reduced, despite the higher ghrelin levels, which suggests that the voracious appetite associated with PWS is related to another mechanism50). Ghrelin may be involved in the instigation of binging and in the hyperphagic stage47).
3. Importance of early weight control
The weight of patients with PWS begins to increase between 18 and 36 months of age, without a significant increase in food intake51), that is, the onset of obesity is observed before a substantial increase in food intake or interest is seen. Therefore, early management of eating behavior is important. At present, the only available control for hyperphagia for most people with PWS is close mentoring by caregivers and life-long restricted access to food, with food cupboards and refrigerators often requiring locks.
Sexual development
1. Hypogonadism
Genital abnormalities, including delayed or incomplete pubertal development and infertility, are common in PWS patients of both genders. Although some female patients with PWS undergo spontaneous menarche, most experience primary or secondary amenorrhea or oligomenorrhea. Most clinicians agree that cryptorchidism in male patients should be corrected to enable the detection of testicular malignancies. Recent findings indicate that hypogonadism in PWS patients is not exclusively hypothalamic and that a primary ovarian or testicular defect contributes to the abnormal reproductive function52,53). At present, the literature contains few reports regarding the use of sex hormone replacement therapy (transdermal or intramuscularly administered testosterone preparations for boys and oral or transdermal estrogen preparations for girls) in affected individuals, although this type of treatment is likely to be beneficial in improving secondary sexual characteristics and helping to prevent osteoporosis. The need for substitution therapy should be assessed individually against the background of the development of BMD, general activity, and quality of life.
2. Early adrenarche and precocious puberty
The normal appearance and development of pubic hair ("adrenarche" or "pubarche") suggest unimpaired adrenal androgen secretion, corresponding to the observed normal androstenedione levels. Adrenarche typically occurs early in many children with PWS13,54). True precocious puberty is very rare in PWS patients and only a few cases have been reported55). Sexual development is incomplete in most PWS patients, and this finding is consistent with arrested pubertal development. In patients with PWS who exhibit precocious puberty, the use of a luteinizing hormone-releasing hormone analog along with GH can permit stature closer to the target height. However, PWS patients with precocious puberty need individualized treatment.
Hypothyroidism
Thyroid axis dysfunction seems to be a frequent feature in infants with PWS. Pediatricians should be aware of this association so this possibility is considered while evaluating PWS patients. Childhood is a critical period of thyroid hormone action on neurological development, and neonatal thyroid-stimulating hormone (TSH) screening is not an accurate tool to diagnose thyroid axis dysfunction56). Published data regarding thyroid hormone levels in PWS children undergoing GH therapy are very limited. Free T4, T3, and TSH levels should be monitored regularly in PWS children, particularly during GH treatment57).
Adrenal insufficiency
Several reports have indicated an estimated PWS-associated mortality rate of 3% yearly17,18,58,59). Disturbances in the hypothalamus-hypophysis-adrenal axis are hypothesized to be responsible for this mortality rate- or at least to represent concurrent factors consistent with an inadequate or late response during infections or relevant dehydration episodes60). Central adrenal insufficiency (CAI) probably results from an inappropriate corticotrophin-releasing hormone secretion by the hypothalamus. In particular, del15 patients, and female, obese, and older patients were reported to be at a higher risk of CAI60). Although clinically relevant adrenal failure in PWS children is rare, glucocorticoid treatment should be considered during intercurrent illness in PWS patients without a recently proven normal adrenal function. Cases of moderate-to-severe stress in all PWS infants warrant replacement treatment with hydrocortisone at 30 to 70 mg┬Ęm2┬Ęday-1, divided into 3 doses60).
Conclusion
In this review, we attempted to summarize the genetic and endocrine dysfunctions in PWS patients and to provide useful information for optimizing the management of PWS patients. This disorder is a disabling condition associated with dysfunction of the hypothalamic-pituitary axis and is mainly characterized by GH deficiency and hypogonadism. Many studies have described the beneficial effects and concerns of GH replacement therapy in children with PWS. Overall, GH treatment, when coupled with a strictly controlled diet in early childhood, may help to reduce obesity, improve neurodevelopment, and increase muscle mass. However, long-term safety studies need to be conducted, particularly with regard to the effects of GH treatment on glucose metabolism and scoliosis. Thyroid hormone levels should be regularly monitored in PWS children, particularly during GH treatment. Glucocorticoid treatment should be considered during intercurrent illness in PWS patients without a recently proven normal adrenal function. In conclusion, a more active approach for correcting these hormone deficiencies would benefit PWS patients.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


