




The published article entitled “Systemic autoinflammatory disorders’’ reviewed the recent molecular mechanism, clinical manifestations, diagnostic approach with molecular genetic testing, and treatment of systemic autoinflammatory disorders (SAIDs) [1]. That article introduced major categories of SAIDs by mechanism, including inflammasomopathies and other interleukin (IL)-1-related conditions, endoplasmic reticulum stress, endogenous antagonist mutations, nuclear factor kappa beta (NF-κB) signaling dysregulation, interferonopathies, and adult-onset SAIDs [1].
SAIDs are a group of rare monogenic disorders characterized by excessive activation of antigen-independent inflammatory pathways due to an imbalance of the innate immune system. The clinical manifestations of SAIDs include periodic fever, a multisystemic inflammatory response, and immunodeficiency [2].
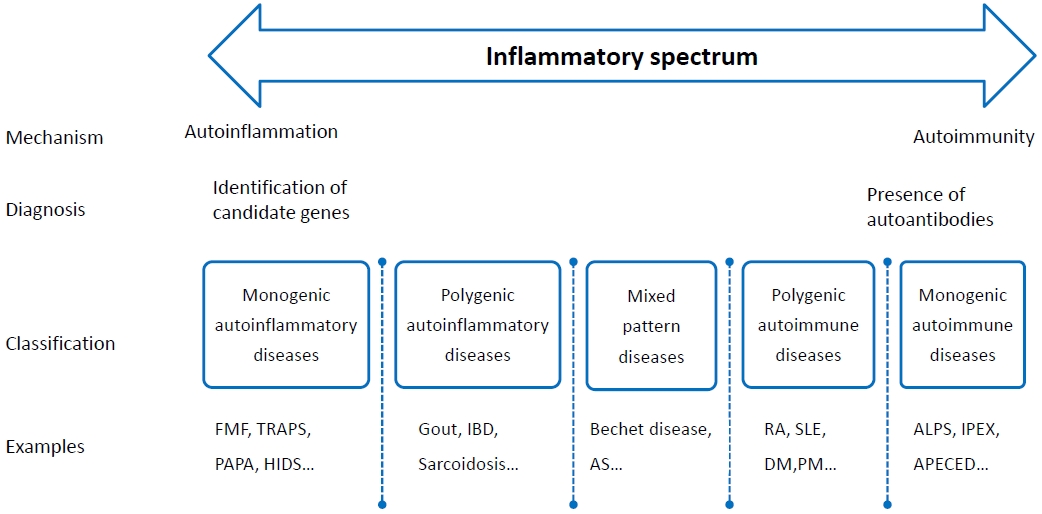
Autoinflammatory and autoimmune disorders are the 2 main categories of immune system disorders. Although these 2 inflammatory disorders are common, they have unique characteristics. In most cases, innate immunity dysfunction indicates the presence of autoinflammatory disorders caused by genetic mutations, while adaptive immunity dysfunction indicates the presence of autoimmune disorders [3].
Discrimination between self and nonself is a fundamental property of the immune system. Self-tolerance is the process by which immune cells become unresponsive to self-antigens to prevent damage to healthy tissues. Self-tolerance failure results in an immune response against self-antigens. Such reactions are called autoimmunity and may cause autoimmune disorders [4]. In autoimmune disorders, lymphocytes become overactive and cytokine release leads to the abnormal secretion of autoantibodies, resulting in inflammation and tissue and organ damage. More than 100 systemic or organ-specific autoimmune diseases have been identified to date. Systemic lupus erythematosus is the most common systemic autoimmune disorder, while type 1 diabetes is an example organ-specific autoimmune disorder [5].
Autoinflammatory disorders are characterized by inflammation due to hyperactivation of antigen-independent immune pathways. Most autoinflammatory disorders are hereditary diseases that develop due to dysregulation of innate immunity through various pathways, including inflammasomes, endoplasmic reticulum stress, NF-κB dysregulation, and interferon production [6]. Overactivation of proinflammatory cytokines mediates and participates in the inflammation characteristic of autoinflammatory disorders.
More than 40 known autoinflammatory disorders can be classified into several categories based on the underlying genetic mutations and clinical findings [7,8]. Monogenic autoinflammatory disorders include familial Mediterranean fever (FMF), tumor necrosis factor (TNF) receptor-associated periodic syndrome, and cryopyrin-associated periodic syndrome. Polygenic autoinflammatory diseases include systemic juvenile idiopathic arthritis and Behçet disease.
FMF is an inflammasome caused by mutations in the MEFV gene, and most other autoinflammatory disorders are caused by NF-κB and/or aberrant TNF activity, interferon production, and excessive IL-1 signaling [8].
Autoinflammatory and autoimmune disorders affect each other; in fact, in many diseases, these 2 inflammatory reactions occur together (Fig. 1).
In SAIDs, the immune system becomes overactive and produces excessive amounts of cytokines, resulting in various symptoms such as periodic fever, joint pain, skin rash, serositis, and lymphadenopathy. The diagnosis of SAIDs is based on clinically suspicious findings, laboratory tests for inflammatory cytokines, and genetic confirmation. As genetic sequencing technology and analysis have improved and costs have decreased, more cases have been identified. By 2022, the United States market will offer over 429 genetic immune dysregulation panels designed to diagnose SAIDs. The goal of SAIDs treatment is to suppress the hyperinflammatory immune state, restore the function of multiple systems, and improve quality of life [2]. Conventional treatment approaches, such as nonsteroidal anti-inflammatory drugs, corticosteroids, and colchicine, have been used, while significant advances have recently been made, including biological agents such as IL-1 blockers, TNF inhibitors, Janus kinase inhibitors, and gene therapy.
In conclusion, SAIDs are a spectrum of innate immune disorders that induce multisystem inflammatory damage through excessive inflammatory pathway activation. An early and accurate diagnosis is important to appropriate SAID treatment and management. However, owing to the rarity of SAIDs, studies on their pathogenesis and treatment are limited; therefore, SAIDs are currently difficult to manage. The incidence of SAIDs will further increase owing to advances in genetic research and biotechnology, leading to their better understanding, diagnosis, and treatment.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


