




Introduction
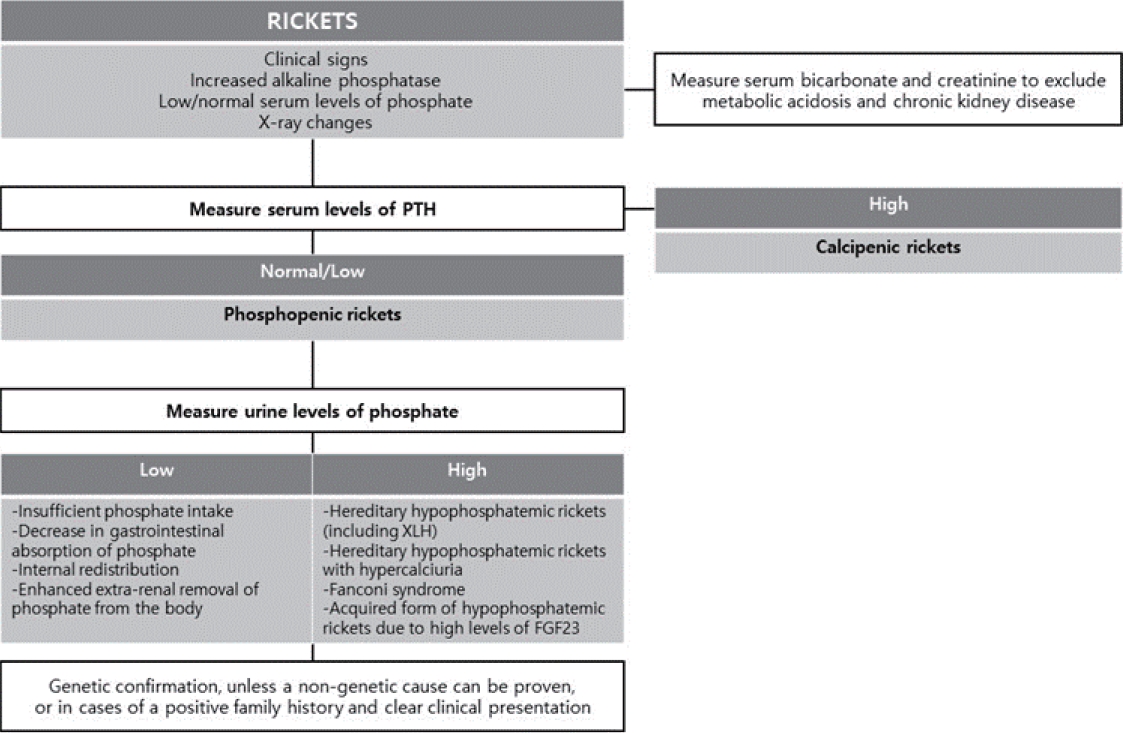
The kidney tubules reclaim the majority of nutrients filtered by glomerular filtration into the urinary space through diverse mechanisms. When the reabsorption of filtered phosphate is impaired by acquired tubular damage or genetic defects in sodium-phosphate cotransporters or their regulators, significant hypophosphatemia occurs. If this persists in children, rickets, a failure to mineralize growing bones, because phosphate is required with Ca to form hydroxyapatite and mineralize the bone [1]. The common cause of rickets is the nutritional deficiency of vitamin D or Ca intake, previously denoted calcipenic rickets, while phosphopenic or hypophosphatemic rickets is relatively rare and often has a genetic cause [2].
X-linked hypophosphatemia (XLH; Mendelian Inheritance in Man #307800) is the most common cause of hypophosphatemic rickets, comprising 90% of familial cases and 70% of sporadic cases and affecting one of every 20,000 people in the general population [2,3]. It was first labeled vitamin D–resistant rickets in 1937, and its inheritance pattern of X-linked disorders was elucidated in 1958. Since female carriers of pathogenic variants are also affected, it is an X-linked–dominant disorder [4]. In 1972, the nature of XLH as an inborn error of phosphate transport was revealed, and the causative gene PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) was discovered in 1995 [5,6]. Although conventional therapy for XLH was introduced approximately 4 decades ago, the temporary replacement of oral phosphate salts and activated vitamin D cannot completely control chronic hypophosphatemia, leaving patients with incomplete healing of rickets and residual skeletal deformity as well as at risk of endocrine abnormalities and adverse drug reactions [7]. However, understanding the pathophysiology has led to the development of a targeted therapy, burosumab, a fibroblast growth factor-23 (FGF23) inhibitor, which was recently approved in Korea for the treatment of XLH. This review provides insight into the diagnosis, evaluation, treatment, and recommended follow-up for a typical case of XLH and details its pathophysiology [8].
Presentation and evaluation
1. Clinical vignette part 1
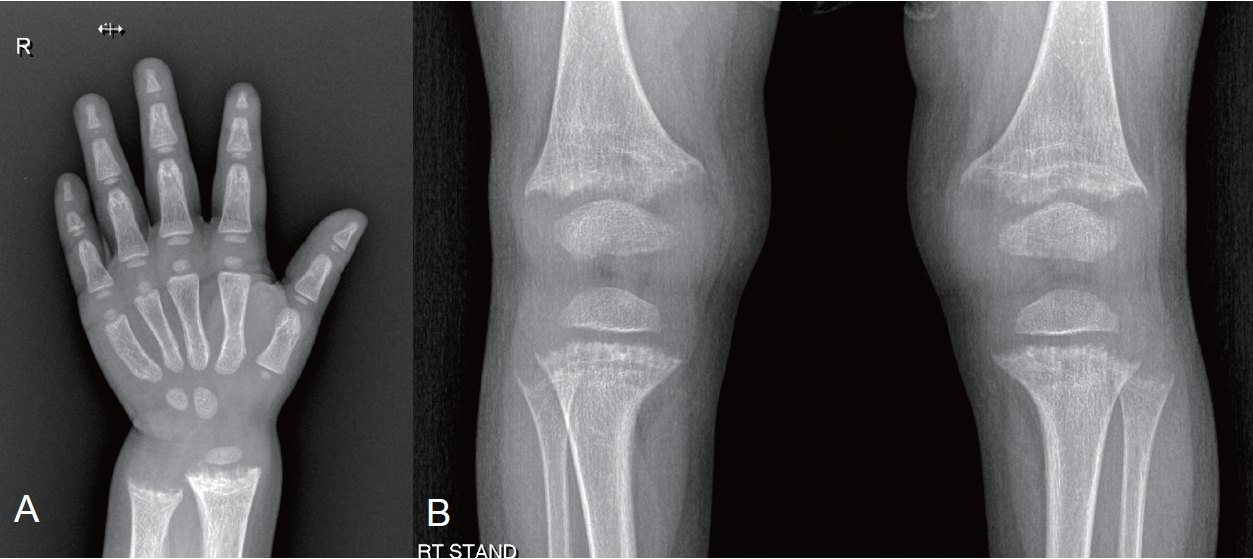
A 25-month-old girl visited the outpatient clinic with growth impairment. Her perinatal and family histories were unremarkable, and she was receiving vitamin D supplementation. Upon examination, her height was 77.6 cm (<3rd percentile), while her weight was 9.8 kg (5th to 10th percentile). Her laboratory test results were unremarkable (serum calcium [Ca], 9.8 mg/dL; serum creatinine [Cr], 0.39 mg/dL) except for an elevated alkaline phosphatase (ALP, 1,087 IU/L) and hypophosphatemia (serum phosphorus [P], 2.4 mg/dL). Radiography of the hand and knee revealed metaphyseal fraying and growth plate widening suggestive of rickets (Fig. 1). An additional workup for rickets showed a normal urine Ca/Cr ratio (0.04) with an elevated urine P level (75.2 mg/dL), low tubular reabsorption of phosphorus (TRP, 69%), and a low ratio of tubular maximum reabsorption of phosphorus to glomerular filtration rate (TmP/GFR, 1.65; reference range, 3.25–5.51). Serum 25-hydroxy (OH) vitamin D and parathyroid hormone (PTH) levels were within the normal ranges (45.47 ng/mL and 67.8 pg/mL, respectively), while the 1,25-dihydroxy vitamin D (1,25(OH)2D) levels was elevated (99.95 ng/mL). The genetic diagnosis of XLH was made by the identification of loss-of-function mutations of the PHEX gene.
XLH usually presents as typical rickets, but its symptoms vary according to age at presentation and disease severity [9]. Short stature, bowing of the legs, difficulty walking from the deformity and weakness (waddling gait), and an unusual shape (dolichocephaly, frontal bossing) of the skull from craniosynostosis are common chief complaints [10-12]. The family history may be significant in X-linked–dominant patterns for disproportionate short stature, leg deformities, dental abscesses, periodontal disease, and hearing loss along with osteoarthritis, enthesopathy, and (pseudo) fractures [2]. Upon examination, typical findings of rickets, namely widening of the wrists and metaphysis and valgus or varus deformities of the legs, are noted. Often, the children of an affected female patient visit the clinic and are diagnosed through laboratory tests before the disease develops. Laboratory characteristics of XLH include hypophosphatemia, normocalcemia, hyperphosphaturia (low TRP and TmP/GFR), and inappropriately normal 1,25(OH)2D levels [13,14]. While normal PTH is a typical finding of XLH, if the patient is taking supplemental vitamin D, PTH and 1,25(OH)2D might be mildly elevated as shown in the vignette.
Pathophysiology
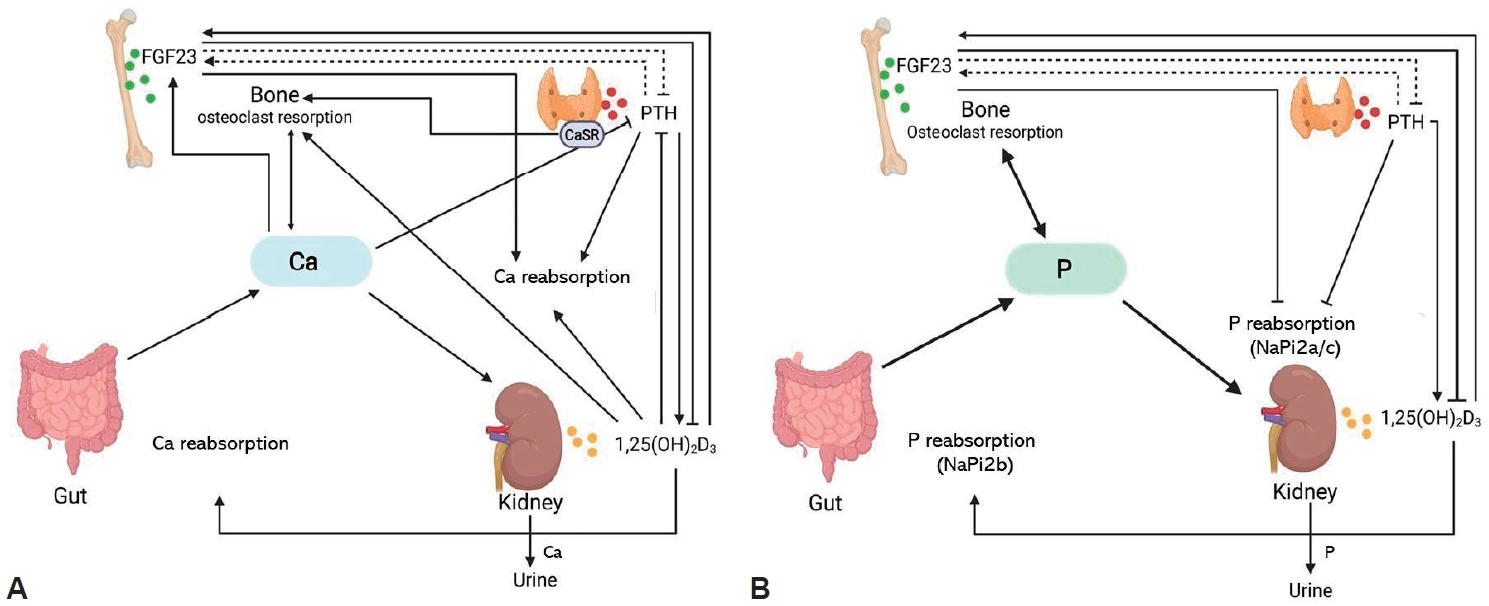
To understand the pathophysiology of XLH, it is necessary to understand the regulatory mechanisms of Ca and phosphate (Fig. 2). When serum P is decreased, its intestinal absorption and tubular reabsorption are increased through the sodium-coupled phosphate cotransporters NaPi2b (intestine), NaPi2a, and NaPi2c (kidney), which are upregulated by increased 1,25(OH)2D production. With hyperphosphatemia, the phosphaturic hormone FGF23 is activated. It then lowers serum P by decreasing renal tubular reabsorption of phosphate through the downregulation of NaPi2a and NaPi2c and suppresses the activity of renal 25(OH)D3 1α-hydroxylase to reduce the production of 1,25(OH)2D [15-19]. In the case of hypocalcemia, PTH is upregulated to increase serum Ca by mobilizing it from the bone, increasing the production of 1,25(OH)2D to absorb/reabsorb more Ca from the intestine and kidney, and downregulating kidney tubular reabsorption of P [20]. In XLH, the FGF23 level is increased regardless of the serum P level, causing phosphaturia and decreased or inappropriately normal 1,25(OH)2D levels, the main pathogenic mechanism in XLH (Fig. 2) [21]. However, which is the mechanism by which FGF23 is increased in XLH has not yet been elucidated [22-24]. Additionally, other regulators of bone mineralization, such as osteopontin and acid serine aspartate–rich, MEPE-associated protein peptides, also increase in XLH [24,25]. On the other hand, such an increase in FGF23 leading to phosphaturia is not unique to XLH. Still, other genetic conditions affecting FGF23 or its regulators have a similar pathophysiology (Table 1, Fig. 3). In recent studies, the detection rate of PHEX mutations was reportedly 33.0%–83.3% when patients with phosphopenic rickets were screened by targeted next-generation sequencing [26-28]. Therefore, a confirmational diagnosis of XLH is usually obtained only after a genetic diagnosis is made.
Differential diagnosis
The typical clinical picture of hypophosphatemic rickets occurs in conditions other than XLH such as dietary phosphate deficiency, including very low birth weight exclusively breastfed infants, infants fed elemental or hypoallergenic formulas, and patients receiving parenteral nutrition as well as impaired phosphate bioavailability due to overuse of P binders and gastrointestinal disorders [2,28-30]. Other conditions, such as primary renal tubulopathies, may resemble XLH, including hereditary hypophosphatemic rickets with hypercalciuria, Dent disease 1, cystinosis, other hereditary forms of Fanconi syndrome, and iatrogenic proximal tubulopathy [30].
Treatment
1. Clinical vignette part 2
The patient was prescribed alfacalcidol 0.25 µg once a day and potassium phosphate/sodium phosphate 125 mg (Phospha 250 Neutral 0.5 tablet) 4 times a day. She will be followed up monthly until her ALP level normalizes, quarterly until 5 years of age, every 3–6 months until puberty, and more frequently during puberty according to the guidelines [31]. Growth, neurological signs, and metabolic control will be monitored for normal ALP, PTH, and normocalciuria. Blood pressure and dental examinations twice a year, renal ultrasonography and an orthopedic examination once a year, and yearly hearing tests from 8 years of age will also be performed. Once available, burosumab, an anti-FGF23, will be prescribed until her growth is complete.
XLH was historically managed with oral phosphate supplementation and activated vitamin D (calcitriol or alfacalcidol) to offset its renal loss. The recommended doses of each medication are listed in Table 2 [31-33]. Once provided with phosphate and active vitamin D, rickets symptoms are usually ameliorated and patients grow better and complain of less bone pain [34-36]. However, the efficacy of such management is often insufficient, as the skeletal deformity progresses and osteotomy becomes necessary in many cases [37,38]. In addition, a large amount of phosphate is necessary to normalize serum Plevels, excessive amounts of which inhibit intestinal Ca absorption, leading to secondary hyperparathyroidism and aggravating bone resorption and phosphaturia [39]. However, an excessive calcitriol dosage may cause hypercalciuria and nephrocalcinosis. Nephrocalcinosis is a common complication of XLH, not from the disease itself but from the replacement therapy [40,41]. Therefore, clinicians need to well titrate and balance the dosage of phosphate supplementation and active vitamin D to maintain normal PTH levels and normocalciuria.
The target of XLH management is the recovery from rickets with the normalization of serum ALP levels but not the normalization of serum P levels. Another problem with classical treatment is poor compliance owing to gastrointestinal problems caused by phosphate supplementation.
2. Anti-FGF23 treatment
As an increase in FGF23 is the main pathogenic mechanism of XLH, the inhibition of FGF23 may be the ideal treatment approach. Burosumab, a fully humanized monoclonal immunoglobulin G1 antibody that neutralizes FGF23, was developed in the early 2000s and showed efficacy and safety in a series of clinical trials in adults and children [42-48]. This targeted medicine increases serum P levels and improves physical function.
In an open-label, phase 2 trial of XLH children, 52 patients aged 5–12 years were randomly assigned to receive burosumab every 2 weeks or every 4 weeks for 64 weeks. Every 2 weeks dosing improved TRP with more stable serum P levels than every 4 weeks dosing and resulted in substantial healing of rickets in nearly all the children with severe disease. These results indicate that the administration of burosumab every 2 weeks is an appropriate regimen for children with XLH [44]. In an active-controlled, open-label, phase 3 trial of XLH children, 61 patients aged 1–12 years of age were randomly assigned to receive burosumab subcutaneously every 2 weeks or conventional therapy for 40 weeks. Rickets severity and height z-score improved significantly more in the burosumab versus conventional therapy group [46]. In another open-label phase 2 trial of XLH children aged 1–4 years of age, 13 patients received burosumab every 2 weeks for 64 weeks. In this study, burosumab increased serum P levels, improved the rickets, and prevented an early decline in height z-score [48].
Additional benefits of this targeted therapy include elimination of the burden of frequent medication dosing and the side effects of conventional medications of gastrointestinal discomfort, secondary hyperparathyroidism, hypercalciuria, and nephrocalcinosis. In clinical trials of XLH children, most patients who received burosumab experienced an adverse effect, but most were mild or moderate in severity with the most common being injection site reactions, hypersensitivity, headache, cough, vomiting and pyrexia [44,46,48]. As a result, burosumab was approved for the treatment of XLH and tumor-induced osteomalacia in patients older than 1 year with radiographic evidence of bone disease in the United States, Europe, and Japan in 2018, and in 2020 in Korea. It is expected to be available in Korea in early 2023 [8].
The dosage of burosumab is usually titrated to achieve a lower age-related normal range of serum P levels [32]. For growing children, its starting dosage is 0.8 mg/kg every 2 weeks subcutaneously (not to exceed 90 mg; after the completion of growth, 1 mg/kg every 4 weeks subcutaneously) and increased by 0.4 mg/kg (up to 2.0 mg/kg) every 4 weeks to raise fasting serum P levels within the lower end of the normal reference range for age. Burosumab should be withheld if the fasting serum P level at 7–11 days postinjection exceeds the upper range of normal and can be restarted at approximately half of the previous dose when the serum P concentration is below the normal range [32]. Assessments of TmP/GFR are recommended to confirm improvement in renal phosphate wasting [31].
Follow-up evaluation
Recommended follow-up intervals and evaluations are listed in Table 3. In addition to disease activity and growth, treatment-associated side effects such as hypercalciuria, nephrocalcinosis, nephrolithiasis, abnormal PTH levels (high or low) in patients receiving conventional treatment, and hyperphosphatemia or hypervitaminosis D (1,25(OH)2D) in patients receiving burosumab treatment can occur [31]. Since XLH is accompanied by complications encompassing orthopedics, neurosurgery, dentistry, and otolaryngology, careful evaluations for such conditions are also needed. Notably, the majority of adult and children with XLH experience bone, joint, and/or muscle pain. Craniosynostosis is observed in about one-third of patients with hypophosphatemia; thus, strict monitoring of head circumference and skull shape during the early years of life is essential. The possibility of increased intracranial pressure, headache, and neck pain from craniosynostosis, a type 1 Chiari malformation, or syringomyelia requires consideration [4,11,12,31,49]. Also, since approximately two-thirds of patients with XLH suffer from dental and periodontal lesions, regular dental examinations to screen for delayed dentition and tooth abscesses are necessary. Hearing problems are rarely seen in children with XLH, but various degrees of hearing loss can occur in adults with XLH, so evaluations for hearing loss starting at 8 years of age are recommended [4,50-52].
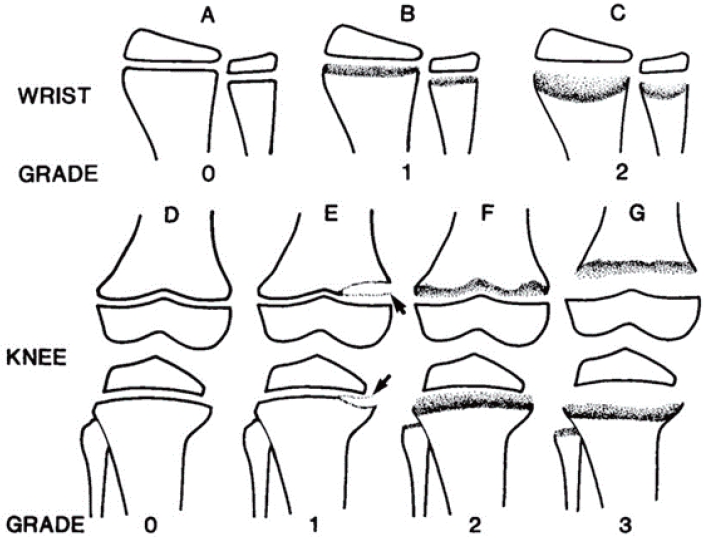
Radiographic evidence of bone diseases can be quantitatively assessed by the Rickets Severity Score (RSS) using radiographs of the wrists and knees (Table 4, Fig. 4) [50,53]. The RSS is a validated measure of rickets severity ranging from 0 (no rickets) to 10 (severe rickets) based on the degree of metaphyseal fraying and concavity and the proportion of affected growth plate affected (https://www.rsstrainingtool.net/).
Prognosis
Once an individual fully matures, their requirement for Ca and phosphate decreases with the spontaneous amelioration of hypophosphatemia, albeit with incomplete resolution. The final height of the XLH patient depends on interventional timing and mode [25,54]. The efficacy of growth hormone treatment is uncertain, especially among those with less optimal metabolic control [54-58]. Residual lower-leg deformities are often debilitating, and about half of affected individuals require surgical correction consisting of corrective osteotomies for those who attain their adult height and epiphysiodesis for those who are still growing [31,37,38,49]. Optimal metabolic control is a prerequisite for surgery. In addition to deformities, osteophytes, enthesopathies (ossification of the tendon), osteoarthritis, (pseudo)fractures, and spinal stenosis may occur, resulting in pain and/or immobility [7,31,59,60]. Supplemental phosphate and active vitamin D may relieve the symptoms [61,62]. Dental care is important for patients with XLH because dental abscesses, poorly mineralized dentin, and periodontitis are common. Although conventional treatment with oral phosphate and active vitamin D may ameliorate this problem, the efficacy of burosumab in this area remains unclear [4,61].
Conclusion
Although rare, XLH is a clinically significant disease whose delayed management may result in short stature and debilitating deformity. Especially since a targeted therapy based on its pathophysiology is now available, the suspicion and early diagnosis of XLH in every case of short stature and hyperphosphaturic hypophosphatemia is necessary.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


