




Infantile-onset multisystem neurologic, endocrine, and pancreatic disease (IMNEPD) is a rare inherited recessive disorder that is caused by biallelic mutation in PTRH2 gene. The first cases were described in 2014 by Hu et al. [1] who exposed the novel disease phenotype, genetic cause, and the functional analysis in human and mice tissues. The main features of IMNEPD are intellectual disability, global developmental delay, deafness and exocrine and endocrine pancreatic insufficiency. The phenotypic variability was reported in individuals carrying the same mutation [2]. Clinical variability was observed and the frequency of neurological abnormalities was found highest. Diverse types of mutations were discovered having the missense mutation the commonest [3]. Studies conducted in the Middle Eastern and Asia region reported varying genetic mutations [1,4-6]. We presented 2 siblings diagnosed with IMNEPD with phenotype and genotype description.
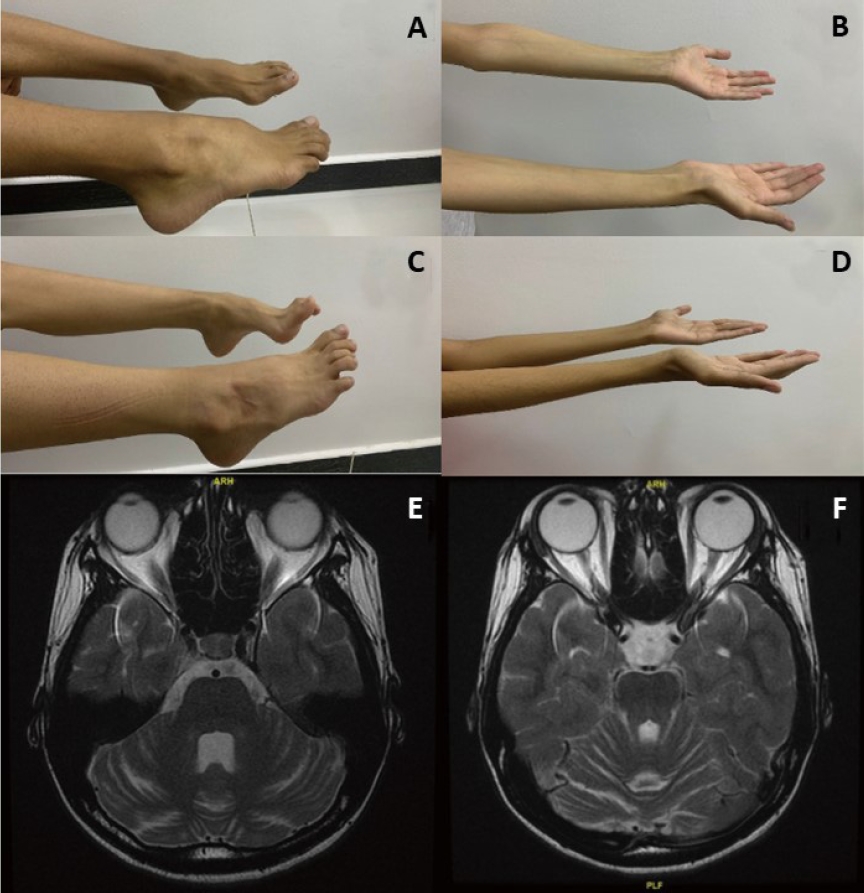
A 17-year-old female born to consanguineous parents (Table 1). Parents noticed global developmental delay at 1 year of age. Patient had difficulties in gross motor skills, walks in a slow gait and severe pes cavus deformity (Fig. 1A). Patient was diagnosed recently with mild scoliosis. In addition to speech delay and learning difficulties, she was hypotonic with muscular atrophy and depressed deep tendon reflexes (Fig. 1B). Electroencephalogram (EEG) at 1 year of age showed mild to moderate generalized nonspecific disturbance of cerebral activity. She received antiepileptic medications at 1 year for couple of years. On follow-up, her repeated EEGs at 2,11 and 13 years of age were unremarkable. Brain magnetic resonance imaging (MRI) recently reported cerebellar atrophy when compared to normal previous MRI (Fig. 1E, F). At the age of 12 years, patient was diagnosed with insulin-dependent diabetes mellitus when hemoglobin A1c (HbA1c) was 15.3% but was not in ketoacidosis. She was referred to genetic clinic and performed whole exom sequencing after written consent. It showed a novel homozygous variant in PTRH2 gene classified as likely pathogenic according to American College of Medical Genetics classification (Supplementary 1).
An 18-year-old male who is brother of 1st case (Table 1). The patient followed almost the same clinical course as his sister. Parents noticed global developmental delay in the 1st year of life. Currently, he has difficulties in gross motor skills, walks in a slow gait and has severe pes cavus deformity (Fig. 1C). In addition to speech delay and learning difficulties, he is hypotonic with muscular atrophy with depressed deep tendon reflexes (Fig. 1D). EEG showed mild generalized disturbance of cerebral activity with infrequent generalized epileptic discharges. Antiepileptic medications were started for couple of years. His repeated EEGs at 4, 12, and 15 years of age were unremarkable. Targeted genetic testing confirmed the diagnosis (Supplementary 1).
Clinical progression and mutations
PTRH2 gene produce a mitochondrial protein which has a functional role in human cognitive function and the regulation of cell growth and survival. It involves in regulation of apoptotic signaling and myogenic differentiation [7]. This explains the progressive nature of the disease. Hu et al. [1] reported an affected girl who became dependent on wheel-chair at age of 15 years due to muscle weakness and ataxia. Our case 1 showed progressive difficulty in walking due to demyelinating neuropathy and scoliosis but she is still ambulatory. She showed progression in the hearing impairment. Another study in Syrian patient (age 17 year) showed IMNEPD due to a novel nonsense mutation in PTRH2 gene (c.324G>A (p.W108*) resulted in limb and truncal ataxia [5]. Supplementary 2 showed the clinical variability of IMNEPD and demonstrated neurological manifestation predominate as PTRH2 was highly expressed in brain tissue. In our patients, we discovered homozygous variant in PTRH2 gene which is the second family to have duplication type. This adds to the postulation that protein-truncating variants is more severe [3,5,6].
Cerebellar atrophy
MRI brain for our first case showed progressive cerebellar atrophy and this has been frequently reported in patients with IMNEPD [1,2,8]. Parida et al. [3] estimated the presence of cerebellar atrophy in 40%. The lack of cerebellar atrophy in some patients with missense mutation is mainly related to residual PTRH2 [2]. In absence of PTRH2, there will be downregulation of the mammalian target of rapamycin (mTOR) signaling and lead to loss of Purkinje cell in cerebellum and progressive cerebellar atrophy [8]. It has been reported that mTOR1 agonists can rescue Purkinje cell and improve mTOR signaling [9]. This raises the hope for a potential therapy.
Diabetes
Our case was diabetic and her affected sibling in prediabetic state. The youngest age when diabetes was discovered was 12 years of age [3]. Another study reported development of antibody negative diabetes (HbA1c value 10.2%) with neuro cognitive impairment and deafness at the age of 13 year emphasized on regular monitoring of diabetes [10].
We herein recommend regular check of glycosylated hemoglobin after the 1st decade of life and encourage to do a neuroimaging at diagnosis and follow-up especially if the patient has neurological manifestation.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


