




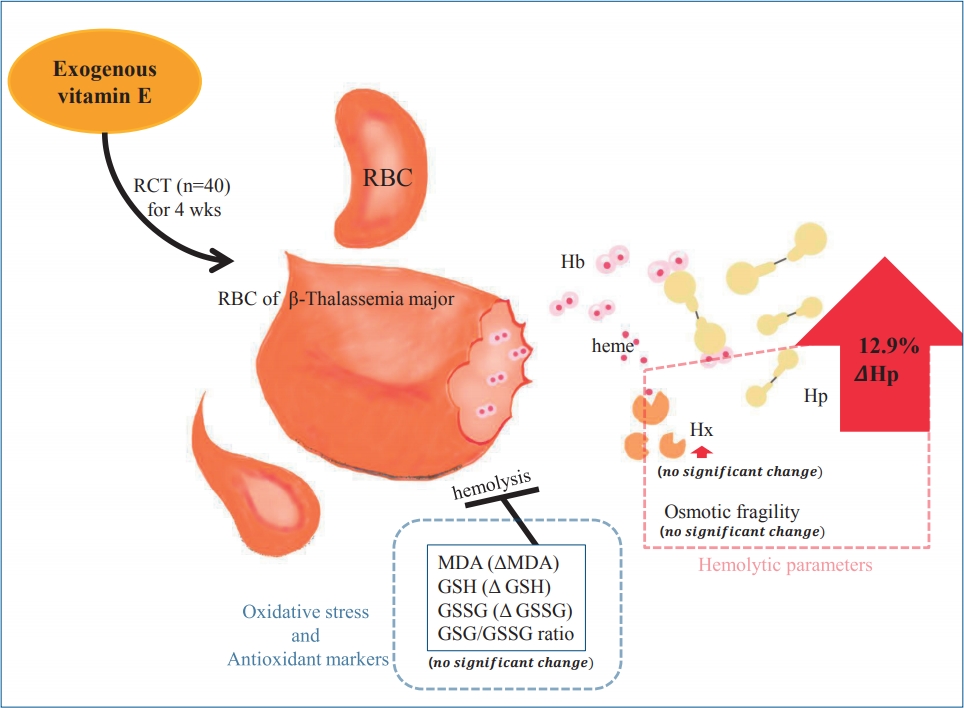
Graphical abstract. Effect of α-tocopherol on hemolysis and oxidative stress markers on the red blood cell (RBC) membranes of patients with β-thalassemia major. Hp, haptoglobin; Hx, hematopexin; MDA, malondialdehyde; GSH, reduced Glutathione; GSSG, oxidized glutathione.
Introduction
Thalassemia is a genetic disease in the form of a disordered synthesis of the globin chain. The number of patients with thalassemia major throughout Indonesia in 2019 was 10.531, and 666 were registered at the Thalassemia Center of the Department of Pediatrics, Faculty of Medicine, University of Indonesia/Cipto Mangunkusumo Hospital (CMH), Jakarta, in 2018, and the number of new patients was around 51 patients. Beta thalassemia major is chronic hemolytic anemia and an autosomal recessive hereditary due to the complete deficiency in the synthesis of β-globin chains. Beta thalassemia major typically requires lifelong blood transfusions [1,2]. The α-chain aggregates could produce reactive oxygen species, leading to oxidative stressinduced red blood cell (RBC) senescence [3].
The basic clinical symptom of β-thalassemia major is chronic anemia, which reduces the number of RBCs and hemoglobin (Hb) content, resulted from hemolysis and a deficiency in Hb production When RBCs are destroyed within the vascular compartment, Hb escapes into the plasma, dimerizes, and is rapidly bound by the serum protein haptoglobin (Hp). Depending on the extent and frequency of hemolysis, Hp may deplete, and free Hb leads to heme synthesis in plasma. Heme is rapidly transferred to hemopexin (Hx), the plasma protein with the highest binding affinity for heme [4,5]. The measurements of Hp and Hx serum are used as hemolysis markers [6,7]. Osmotic fragility tests can also measure the lysis of RBC as a function of osmotic stress [8].
Data have shown that the indicated lipid of membrane peroxidation in the RBCs of patients with β-thalassemia major is induced by oxidative stress [9]. Reduced glutathione (GSH) is one of the most effective endogenous antioxidant mechanism, turning oxidized to oxidized glutathione (GSSG) and recycling it to its reduced form by glutathione reductase (GR) [10,11].
Consuming vitamin E (α-tocopherol) as an antioxidant can prevent some of the damage in the thalassemia RBC membrane [9,12]. It has been postulated that the low serum of α-tocopherol can be caused by the secondary consumption of the antioxidant in β-thalassemia major patients [13]. There are different guidelines on antioxidant supplementation for the β-thalassemia major in children. The Thalassemia International Federation (TIF), did not recommend, supplementation of α-tocopherol for children with β-thalassemia major [14]. The standard care of thalassemia from Oakland Hospital for the α-tocopherol antioxidant supplement suggested an evaluation of annual nutritional laboratory testing of α-tocopherol [15]. The Indonesian Pediatric Society Coordination work unit from the Hematology Oncology Division recommended α-tocopherol with the doses of 200 mg daily for children below 5 years old, and 200 mg twice daily for children aged 5 years old as the antioxidant supplementation in children with β-thalassemia major [16]. This study, evaluated the efficacy of the α-tocopherol on hemolysis and oxidative stress in RBCs membrane of β-thalassemia major patients.
Methods
This study was a double-blind, randomized clinical trial. The subjects were β-thalassemia major patients hospitalized in thalassemia ward at CMH between December 2016 and July 2017.
1. Study participants
The inclusion criteria were the children aged 5−18 years old who received transfusions in once a month and iron chelation, with no other hematologic disorders, and do not consume any other antioxidants or herbal supplements. Exclusion criteria were β-thalassemia major patients with the acute or chronic infection including hepatitis B or hepatitis C, splenectomy, liver failure and abnormality level of lipid test.
2. Study groups and randomization
The sample size was determined by independent sample t test for examining a difference between two means. Subjects were selected by the consecutive sampling of β-thalassemia major patients and were randomized using block allocation. The patients were randomized into two groups, the placebo and the α-tocopherol group.
3. Intervention
The α-tocopherol dosage age groups were adjusted by the body weight, as suggested based on tolerable upper supplementary intake level by the Institute of Medicine. The range of dosage for children aged 5−8 years old is 200 mg/day, 9−13 years old is 400 mg/day and 14−18 years old is 600 mg/day for four weeks. The supplementation of the α-tocopherol and placebo were prepared by CMH Pharmacy division. The α-tocopherol supplement therapy started immediately after the subjects had finished the regular blood transfusion. Informed consent was obtained from all parents/participants, and this study was approved by the Ethical Committee of the Medical Research, University of Indonesia (No 1050/UN2.F1/Ethic/2015).
4. Clinical procedure and laboratory investigation
Blood samples were collected from each subject just before they received their regular blood transfusions. Clinical laboratory examinations, including complete blood count, alanine transaminase (ALT), total cholesterol, iron profile (serum ferritin, serum iron), and all other hematological parameters tests were performed at the clinical pathology laboratory of CMH.
Hemolysis markers (Hp and Hx serum level) were measured using Human Haptoglobin and Human Hemopexin kit products by Cloud Cone Corp. (Katy, TX, USA) by ELISA method at Terpadu Laboratory, Faculty of Medicine, University of Indonesia.
Plasma malondialdehyde (MDA) level was obtained at Biochemistry Laboratory of the Faculty of Medicine, University of Indonesia. MDA levels were determined by employing the thiobarbituric acid reaction substance (TBARS) method. The GSH, GSSG, and GSH/GSSG ratio were measured using Microplate Assay for GSH/GSSG kit (Oxford Biomedical Research Inc., Metamora, MI, USA). This antioxidant marker was obtained at Biochemistry Laboratory, Faculty of Medicine, University of Indonesia. The analysis of the α-tocopherol levels was measured at the SEAMEO-TROPMED Nutrition Laboratory, Faculty of Medicine, University of Indonesia using the high-performance liquid chromatography method.
5. Study outcome
This study aimed to evaluate the effects of α-tocopherol in hemolysis and oxidative stress marker on the RBCs membrane in β-thalassemia major.
6. Statistical analysis
All parameters were analyzed using IBM SPSS ver. 22 (IBM Corp., Armonk, NY, USA) The t-test was applied to assess the mean difference between the α-tocopherol and placebo groups in a normally distributed data, and the Mann-Whitney test used if the data were not normally distributed. The mean difference between the two groups was statistically significant (P<0.05).
Results
1. Enrollment of study participants
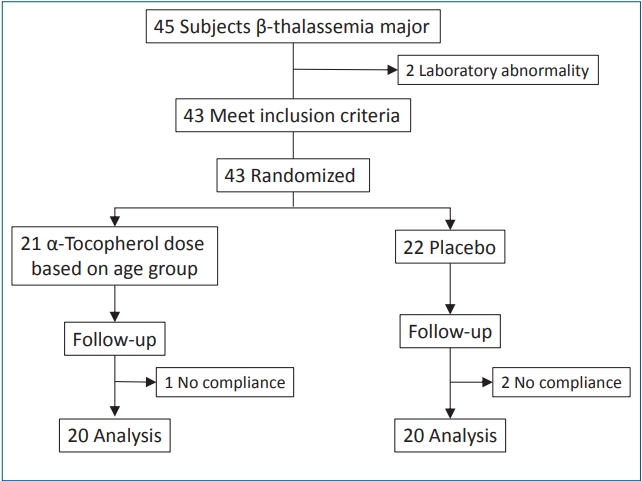
Forty-three patients met the inclusion criteria, but 3 patients did not continue the study because of the failed to comply with the study protocol which is need only 1 month observation. The majority of the thalassemic patients in our center were 2 weeks interval of transfusion, and this became a problem for us for recruiting more subjects. The final subjects were 40 patients, 20 subjects in the placebo group and 20 subjects in the α-tocopherol group (Fig. 1).
The characteristics of participants in the 2 study groups, including age, sex, nutritional status (determined by mid-upper arm circumference measurement of World Health Organization criteria), clinical laboratory, hemolysis, oxidative stress, and antioxidant variables before the intervention are presented in Table 1. There was no significant difference in the subject characteristics between the 2 groups.
2. The effect of α-tocopherol on hemolysis marker
The results of median Hp serum was significantly different between the placebo and the α-tocopherol groups (1.08 mg/dL and 3.01 mg/dL, respectively; P=0.021). The median relative delta of Hp level also showed a significant difference between the 2 groups. It was decreased (-44.15%) in the placebo group, while it was increased (12.09%) in the α-tocopherol group (P=0.042) (Table 2). The mean level of Hx serum after the intervention showed no difference between the two groups (194.54 mg/dL and 180.84 mg/dL, respectively) (Table 2). After the treatment, the mean of RBCs fragility and the mean of delta RBCs fragility also showed no significant difference between the two study groups (Table 2). The result of hemolysis marker in this study indicated that the α-tocopherol, as an antioxidant, was significantly effective in improving the hemolysis by increasing the Hp level indirectly.
Discussion
Our data demonstrated 40 subjects who had completed this study, divided into the placebo and the α-tocopherol groups. The results showed no clinically significant difference between the 2 groups in term of the subject’s characteristics, hemolysis and oxidative stress marker before the treatment. The mean interval of transfusion was 28 days in all of the subjects, with no significant difference between the 2 groups. It is clinically important showed that the mean level of Hb in both of groups still below the Hb pre-transfusion target, which is 9−10.5 g/dL as recommended by TIF [14]. Serum ferritin and saturation of transferrin were also higher than the standard value, and after intervention showed no significant difference between the 2 groups.
The mean level of Hp as a hemolysis marker showed a significant increase in the α-tocopherol group, although the Hp level did not reach the normal limit (P=0.04). Thus, it suggested meaningful implications for the therapeutic use of the α-tocopherol for oxidative stress on the RBCs membrane, which can lead to progressive hemolysis. This data demonstrated that the α-tocopherol could improve hemolysis by the increasing of Hp level.
As the plasma Hp level is decreased in hemolysis as well as in the presence of ineffective erythropoiesis, its depletion is a reliable marker for the instant diagnosis of accelerated RBCs destruction. In β-thalassemia, abnormally high levels of oxidative stress account for accelerated senescence and increased the destruction of erythrocytes [3]. Ragab et al. [17] stated that thalassemia children had more severe Hp depletion compared to healthy subjects and all of the thalassemic children had their Hp level less than 12 mg/dL. Similar to our study, hemolysis and ineffective erythropoiesis in β-thalassemia major leads to Hp depletion and related to the severity of hemolysis.
The normal value for hemopexin level in children is 77 mg/dL (range, 66–100 mg/dL) in average [4]. Both of groups showed an increased average level of Hx serum than normal value after the treatment (194.54 mg/dL and 180.84 mg/dL) in the placebo and the α-tocopherol group respectively, and no significant difference was found. Hemopexin can bind to heme as a product of free Hb from hemolysis process with high affinity [5]. Decreased levels of Hx have been noted in chronic and severe hemolytic states. On the contrary, Hx levels can increased in the acute-phase response [18], from our data there were no increased of leukocyte count in both of 2 groups and we didn’t identify other factors associated with elevated of Hx levels involved in the inflammation process.
Cutillo et al. [19] demonstrated that out of 45 thalassemia major cases, Hp level was absent (Hp 0) in 13 subjects and Hx level was also absent (Hx 0) in 37 subjects. It has been stated that Hx depletion depends on the high concentration of free heme and decreased or absent of Hx also leads to very severe hemolysis. However, there are few data available for patients with β-thalassemia.
In this current study, we found no significant decrease in the Hx level. This finding may be influenced by the role of Hp-free and Hb complex. Therefore, Hp can protect Hb form oxidative modifications that would otherwise prevent its results in the release of free heme to the circulation [20]. We assumed that the α-tocopherol has indirectly benefited for increasing of Hp level by neutralizing lipid peroxidation and preventing hemolysis. Unlike other studies, we found a higher mean of the Hx level than the normal value. The increasing Hx level may be due to the Hx as an acute-phase reactant in the inflammatory process, but we did not investigate the inflammation marker and viral load. Hence, it should be examined in the future research.
The percentage lysis of RBCs was slightly decreased in the α-tocopherol group, but there was no significant difference between the two study groups. In thalassemia patients with transfusion-dependent, the RBCs membrane integrity appears to be destroyed, with the loss of essential polarity and fluidity gradient and oxidative stress by lipid peroxidation may induce membrane fragility and hemolysis [21]. We suggested that the α-tocopherol is considered to be able to protect RBCs membrane and reduce hemolysis.
After the treatment, the MDA level showed no significant difference between the groups, we assumed that influenced by only short-term period of study and oxidative stress still exist. MDA, the product of lipid peroxidation, is generated in excessive amounts, supporting the fact that large quantities of membranebound iron are present in thalassemia RBCs [22,23].
Several studies have shown that MDA is a marker to determine the existence of oxidative stress [24-26]. Mahjoub et al. [9] and Das et al. [27] stated that MDA level decreased significantly in thalassemia patients who received the α-tocopherol. We found different results in our study. The contrary finding may be due to the different subjects’ characteristic, sample sizes, and study protocols.
The ratio between endogen antioxidant (GSH), GSSG, and GSH/GSSG also indicated no significant difference between the 2 study groups. GSH is oxidized to form GSSG, which can be reduced to GSH by the NADPH-dependent GR [28]. The GSH/GSSG ratio reflects the oxidative state and it interacts with redox couples to maintain the appropriate redox balance in the cells [29]. Therefore, the lower ratio of GSH/GSSG may indicate higher oxidative stress in cells [10]. Mahdi [30] and Qaiser et al. [31] reported the lower levels of GSH in β-thalassemia major compared to healthy subjects (P<0.01). This study also revealed no significant difference in GSH, GSSG and GSH/GSSG ratio between 2 study groups.
This study revealed that there was a decrease in the α-tocopherol level in thalassemic children before the treatment. Previous studies showed low levels of the α-tocopherol which can be explained by the excessive iron fraction generated by a lipid peroxidation process with subsequent antioxidant consumption [22,32,33]. In this study, we did not performed a special form for recording the clinically adverse event. Moreover, there was no case reported by the subjects in this study, such as the disturbance of gastrointestinal and bleeding due to oral supplementation of the α-tocopherol.
1. The suggestion of the α-tocopherol mechanism on hemolysis and oxidative stress in RBCs membrane β-thalassemia major
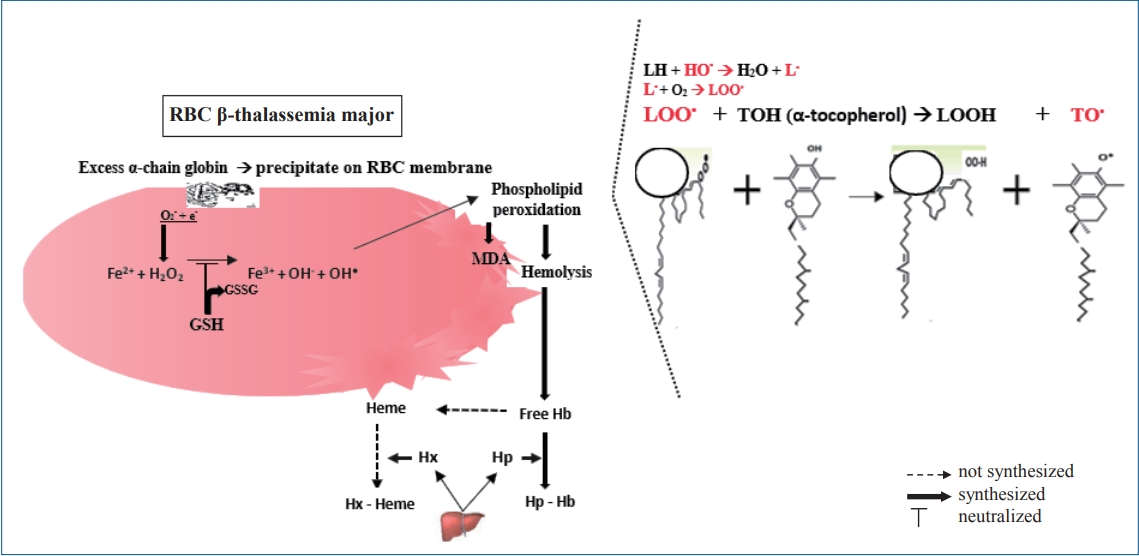
In this study, we proposed the mechanism of GSH and α-tocopherol to neutralize free radical, reducing hemolysis and oxidative stress on the RBC membrane by (Fig. 2): (1) Endogen antioxidant (GSH and GSSG): the reaction of GSH and hydrogen peroxide (H2O2) can inhibit hydroxyl radical (OH●) as oxidant molecule. (2) Exogen antioxidant (α-tocopherol): the α-tocopherol serves as a chain breaker and neutralizes lipid peroxyl radical during the lipid peroxidation in RBCs membranes. (3) Alpha-tocopherol may neutralize hydroxyl radicals (OH●), but it assumed that α-tocopherol is not an efficient scavenger for hydroxyl radicals.
In conclusion, the α-tocopherol as an antioxidant has an important effect in RBCs membrane for reducing hemolysis, by indirectly increasing the level of Hp in β-thalassemia major. Endogenous antioxidant was not affected by α-tocopherol supplementation.
2. Suggestion
Further research using similar study protocols is necessary to investigate the hemolysis marker and oxidative stress for those having transfusion every 2 weeks and pretransfusion Hb between 9 to 10.5 g/dL for longer study period. We also suggest a study to identify marker inflammation in case with elevated of Hx in thalassemic patient.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


