


Introduction
Short chain acyl-CoA dehydrogenase deficiency (SCADD) is a very uncommon disease that is associated with abnormal oxidation of beta-oxidized fatty acid of the short chain acyl-CoA dehydrogenase involved in energy metabolism of fatty acids in the mitochondria. It occurs in one in every 40,000 to 100,000 people, and is associated with various clinical symptoms1,2). It is an autosomal recessive disorder which was first reported in 19873).
SCAD promotes the dehydrogenation of butyryl-CoA (C4-CoA), which is the first step of β-oxidation of short chain fatty acids1,2). The acyl-CoA dehydrogenase (ACADS) gene is a 13-kb-long gene that is located on the long arm of chromosome 12 (12q22), and consists of 10 exons and 1,236 nucleotides. Mutation of this gene leads to SCADD. This results in the production of butyrylcarnitine (C4-acylcarnitine, C4-C), butyryl glycine, butyrate and ethylmalonic acid (EMA). The levels of C4-C is measured in blood, while EMA is measured in urine. Elevated levels of C4-C and EMA are used as biochemical markers for the diagnosis of SCADD2). Clinically, SCADD is associated with hypoglycemia, nausea, growth and development problems, tachypnea, and reduced muscle tone, as well as progressive muscle weakness, growth delay, seizures, microcrania, and scoliosis4). Early diagnosis and treatment is important for the prognosis of metabolic disorders. Compared to those who are diagnosed clinically, patients who are diagnosed in through neonatal screening tend to show normal growth and development5).
Recently, tandem mass spectrometry is being used for the early neonatal diagnosis of metabolic diseases which allows for early treatment for patients to have symptomless metabolic diseases. In this study, we report a case of an asymptomatic patient who was diagnosed with short chain acyl-CoA dehydrogenase deficiency through neonatal screening using tandem mass spectrometry, who was found to be compound heterozygous for the ACADS gene mutation.
Case report
The patient was a male born at 38 weeks and 1 day of gestation. The birth weight was 2,900 g. He was the second child, who was born by caesarian section. There was no significant medical history of the mother. He was also doing well after birth. The patient underwent neonatal screening using tandem mass spectrometry at day 4 after birth, and was found to have elevated levels of butyrylcarnitine at 2.25 µM/L (reference range, <0.99 µM/L). So, he was referred to our hospital for further evaluation.
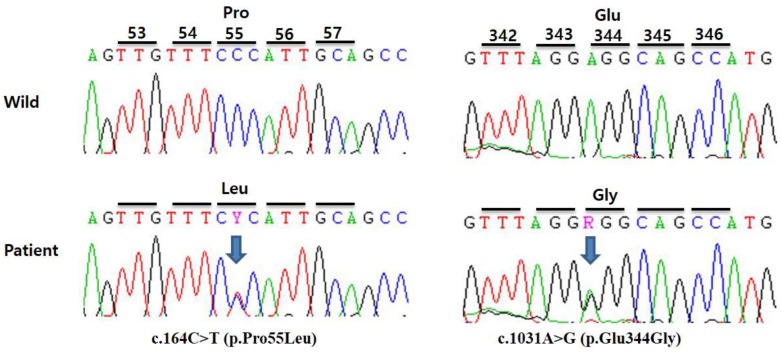
His vital signs were within normal limits at the time of presentation, and there were no abnormal findings in the head, chest, and abdominal examinations. There were no significant abnormalities found in the serum amino acid analysis, but he was found to have ethylmalonic aciduria on urine organic acid analysis, with ethylmalonic level of 87.27 µg/mg Cr (reference range, <0.65 µg/mg Cr). His blood glucose levels, pH, and electrolytes were all within normal limits, which are expected to be abnormal in patients with metabolic disorders. In order to confirm the diagnosis of short chain acyl-CoA dehydrogenase deficiency, we conducted genetic testing. DNA sequence analysis showed that the patient had heterozygous for the mutations in c.164C>T (p.Pro55Leu) on exon 2 and c.1031A>G (p.Glu344Gly) on exon 9, and both of which have been already reported mutations of SCADD6). However, we could not analyze the ACADS gene of his parents because they did not want to know it.
Based on the diagnosis of short chain acyl-CoA dehydrogenase deficiency, the patient received oral vitamin B, L-carnitine from day 20 after birth. The parents were educated on the risks of hypoglycemia with prolonged fasting, and the patient's progress was monitored with regular urinalysis for organic acids. At 4 months of age, the patient's height was 66.6 cm (75th percentile), weight was 7.4 kg (50th–75th percentile), and head circumference was 42 cm (50th–75th percentile). He had normal muscle tone, and was attempting to flip. He had normal head movement, with normal growth and slightly delayed development. Since then, he has no neurological abnormalities found until present, at the age of 35 months. His body measurements at 24 months of age were weight of 11.3 kg (25th percentile), height of 87.5 cm (50th–75th percentile), and head circumference of 49.1 cm (50th–75th percentile). Bayley scales of infant development-2 conducted at 26 months of age showed chronological age (26 months), mental age: 21 months and motor age: 22 months in the developmental age, and social maturity scale (SQ score) of 85.32. The patient's language ability and combined language proficiency age was 2 years and 1 months, which was similar to that expected of his age. The patient received vitamin B, L-carnitine for the above condition, and since 15 months onwards, is only taking L-carnitine.
Discussion
SCADD is an autosomal recessive genetic metabolic disorder caused by the deficiency of SCAD, one of the mitochondrial enzymes involved in the oxidation of fatty acids4). Most individuals identified through newborn screening and affected relatives have been asymptomatic. However, various symptoms have been reported in some patients with SCADD, most frequently developmental delay, seizure, ketotic hypoglycemia, hypotonia, fatigue, failure to thrive, recurrent vomiting, and metabolic acidosis5). Most symptomatic patients with SCADD have presented with predominantly neurologic manifestations, unlike the other β-oxidation defects, via direct neurotoxic effect of increased EMA5). But, these findings are nonspecific and can be frequently observed in other inherited metabolic disorders.
In order to diagnose fatty acid oxidation disorders, tandem mass spectrometry is used to assess the acylcarnitine profile. This tandem mass spectrometry for neonatal screening was introduced across the world from 2000, and became available in South Korea from 20027). This has allowed for screening of asymptomatic patients8). SCADD is characterized by mutation of the ACADS (OMIM #606885) gene on 12q22 of the long arm of chromosome 12. This gene is approximately 13 kb long, consisting of 10 exons and 1,236 nucleotides5,9). There are approximately 70 different types of ACADS mutations6). In Europe, 2 common variants (c.511C>T (Arg147Trp) and the c.625G>A (Gly185Ser) were reported as polymorphism10,11). Each variants accounts the 3%–8% and 22%–43% of normal population, respectively. But, it is important that the homozygosity for one of the polymorphisms is even associated with an increased incidence of elevated EMA excretion5).
There have been reports of asymptomatic SCADD identified in neonatal screening due to G108D mutation in Japan8). In Korea, between 2000 and 2012, the children with developmental delay and mental retardation were evaluated and screened for metabolic and endocrinologic problems, and discovered 3 cases of 508 children as SCADD12). But, these cases were not tested with genetic analysis. Three cases with newborn-screening and ACADS gene confirmed SCADD have been reported in Korea (Table 1)13,14,15). Kim et al.13) diagnosed the first case of asymptomatic SCADD in a female infant diagnosed through neonatal screening with liquid chromatography tandem mass spectrometry, with the identified homozygous mutations of c.1031A>G (p.E344G) on exon 9. In our case, the patient was confirmed to have compound heterozygous mutations of c.[164C>T];[1031A>G] (p.[Pro55Leu]:[Glu344Gly]) on exon 2 and 9, respectively (Fig. 1). Our allele, c.1031A>G, was same as Kim et al.13)'s case and another allele, c.164C>T, was same as Park et al.15)'s report. Cheon et al.14) demonstrated a case with novel mutation of ACADS gene, c.277C>A (p.Leu93Ile) on exon 3 and c.682G>A (p.Glu288Lys) on exon 6. However, they did not analysis their functional study of novel mutations.
There are no set guidelines for the treatment of SCADD until now, and moreover, there is not consensus on the need to treat SCADD. Chronic management of SCADD has been a diet similar to other fatty acid oxidation disorders, consisting of reduction of fat intake to 25% of calories from fat, with smaller, more frequent meals to avoid reliance on β-oxidation5). However, well-tolerable patients with SCADD do not need these strict diets. In episodes of acute metabolic acidosis, intravenous hydration with a solution containing 10% glucose should be used to reestablish an anabolic state, followed by reintroduction of the patient's usual diet. In a few cases, patients have been treated with L-carnitine and riviflavin1). And genetic counseling is recommended for patients and their families.
In this case, the patient received oral riboflavin and L-carnitine daily, and the patient's parents were given dietary education to prevent prolonged fasting for decreasing catabolic state as well as other fatty acid oxidation diseases. Oral supplementation of L-carnitine helps EMA to produce butyrylcarnitine for excretion. Although there is insufficient evidence to supports its use as a treatment, both rivoflavin and L-carnitine are being considered for potential treatment for SCADD patients.
In summary, we have discussed an asymptomatic patient with short-chain acyl-CoA dehydrogenase deficiency who was detected through neonatal screening and is showing normal growth and development to date.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


