


Introduction
Glucose transport 1 (GLUT-1) deficiency is caused by a decrease in the transfer of glucose to the brain due to mutations in the glucose transporter 1 (SLC2A1) gene1). De novo mutations have been identified in the SLC2A1 gene, which consists of 10 exons and 9 introns and contains 492 amino acids in 12 transmembrane domains. Several hot spots have also been reported, including N34, G91, R126, R153, and R333 identified in exons 2, 3, 4 and 8 in the SLC2A1 gene1,2). The pathogenicity of genetic mutations is not well understood; however, previous studies have suggested that an interaction between amino acids influences stabilizing protein structures, and its alteration in mutant results in destabilizing protein-protein interactions and drives structural and functional changes3,4). These mutations in SLC2A1 cause dysfunction of glucose transport to the brain and result in various clinical symptoms. The phenotype extends from mild cases, with only movement disorders, to severe cases characterized by intractable epilepsy with delayed development. Classic phenotypes include epileptic encephalopathy, developmental delay, acquired microcephaly, and complex movement disorders including dystonia, ataxia, and spasticity5). Nonclassic phenotypes include movement disorders without epilepsy or delayed development. Missense mutations are usually related to mild to moderate phenotypes6,7) and to mild mental retardation1). However, most phenotypes are correlated not with the genotype but rather with the degree of glucose transport into the brain1). We described a case who presented with infantile spasms as a rare seizure type of a GLUT-1 deficiency.
Case report
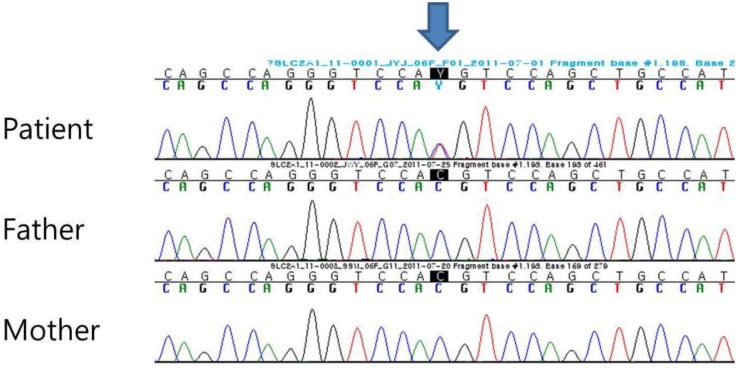
A 13-month-old female patient visited our clinic with delayed development and infantile spasms occurring since 6 months of age. She was born to healthy nonconsanguineous parents at a gestational age of 40 weeks. Her birth weight and head circumference were 3.0 kg and 34 cm, respectively, and were normal for her birth age. At 6 months of age, her seizures developed as flexor spasms with 3–5 clusters and 10–20 spasms per cluster and continued despite antiepileptic drug treatment. Although initial developmental milestones were normal, her development after the onset of seizures regressed to a 3-month-old level, showing the difficulty of turning and crawling at the age of 13 months. On physical examination, she was 79.7 cm in height, and 9.4 kg in weight, and had a head circumference of 42 cm with acquired microcephaly. Her MRI showed normal findings and electroencephalography (EEG) showed very high voltage, disorganized slow waves with multifocal spike suggesting hypsarrhythmia. Her metabolic screening tests, including tandem mass, blood amino acid, lactic acid, pyruvic acid, and urine organic acid, were found to be normal. On cerebrospinal fluid (CSF) examination, after fasting for four hours, her glucose level was low at 39 mg/dL compared to blood glucose at 103 mg/dL with a ratio of 0.38. This finding suggested a GLUT-1 deficiency; mutational analysis of SLC2A1 was performed to confirm the diagnosis. Upon mutational analysis, genomic DNA was extracted from peripheral blood leukocytes and all coding exons and the flanking intronic regions of the SLC2A1 gene were amplified. Cycle sequencing was performed using a BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems, Foster City, CA, USA) and an ABI 3130xl Genetic Analyzer (Applied Biosystems). Bidirectional sequencing of the SLC2A1 gene revealed a de novo heterozygous missense variation (c.1198C>T) at codon 400 (p.Arg400Cys). Further analysis of the parents did not show the p.Arg400Cys variation, indicating that it was a de novo mutation (Fig. 1). After the diagnosis of GLUT-1 deficiency, she was started on a 4:1 ratio ketogenic diet as treatment for GLUT-1 deficiency. After 1 month, her seizures were improved, and the developmental milestones were improved slightly. Improvements in developmental milestones included improvements in turning and clawing, although she did not reach normal development. Six months after starting the ketogenic diet, she started to taper off the antiepileptic drugs. However, at the age of 2 years, her EEG again showed hypsarrhythmia. The record for her ketogenic diet was maintained by the parents. They reported having carrots at every meal for 2 months and not being aware of her seizures. One month after removing carrots from her diet and educating the parents as to the precise ketogenic diet, her EEG showed a disorganized background dominated by theta activity. She discontinued the antiepileptic drugs and changed to a 3:1 ratio of the ketogenic diet at the age of 3 years and 5 months, and she has been maintained on a 2:1 ratio ketogenic diet since 4 years and 5 months of age. Upon developmental testing at the age of 5 years, she showed persistent delayed development with slight improvement, testing at an 11-month-old level on the cognitive test and a 15-month-old level on the performance test.
Discussion
GLUT-1 deficiency is a rare syndrome caused by mutations of the SLC2A1 gene that have an influence on the degree of glucose transport to brain and are associated with various clinical phenotypes. Seizure, the main clinical feature, usually develops during infancy and presents as brief and subtle myoclonic jerk and focal seizures, evolving into a mixture of different types such as generalized tonic-clonic, absence seizure, myoclonic and complex partial seizure8,9). Infantile spasms have been rarely reported as infantile seizure of GLUT-1 deficiency. Our case showed no response for antiepileptic medication and was evaluated in metabolic screening to identify other etiology for infantile spasms. Her CSF finding showed hypoglycorrhachia that was suspected to be the result of GLUT-1 deficiency with confirmation of mutational analysis. She started a 4:1 ratio ketogenic diet as the treatment of GLUT-1 with rapid effect for seizure control and with improvement of development. However, her EEG was aggravated due to short term disruption of ketogenic diet although she did not suffer from infantile spasms for 6 months. Her EEG was improved after the precise ketogenic diet and she stopped taking antiepileptic drugs. Previous study reported ketogenic diet in GLUT-1 deficiency resulted in seizure freedom in 67% patients including one case presenting as infantile spasms and 83% of the patients with seizure freedom had ketogenic diet alone after not taking preexisting antiepileptic drugs9). Ketogenic diets as first treatment of GLUT-1 deficiency provide the improvement of disease condition by supplying an alternative fuel into the brain6). Thus, ketogenic diets should be maintained at least into adolescence although patients must not suffer from epilepsy
Our patient showed a de novo heterozygous missense variation (c.1198C>T) at codon 400(p.Arg400Cys) of exon 9. Exchange of the arginine 400, identified on the SLC2A1 gene in exon 9, is related to reduced glucose transport activity by an inward facing conformational change that alters the interaction of acidic and basic residues4,10). Transmembrane domain 4 (TM 4), the most frequent mutation in GLUT-1, is encoded by SLC2A1 exon 4. Alteration of the intracellular charges at TM 4, including mutations at residues 124,126,130,137, and 146, can have a critical influence on the structure of GLUT-13). The alteration of the structures of GLUT-1 cause dysfunction in the transport of glucose to the brain, and the degree of transport dysfunction is related to various clinical symptoms. So, we compared the clinical symptoms between cases with mutations in exon 9 and cases with all mutations to find clinical differences according to exon site. Including the reported studies and our case, the mutation in exon 9 (n=8) showed clinical symptoms including early onset seizure (75%), microcephaly (71.4%), movement disorder (100%), and pyramidal sign (71.4%),whereas the clinical symptoms in cases with mutations across any of the exons (n=51) included early onset seizure (60.8%), microcephaly (38.6%), movement disorder (80%), and pyramidal sign (36.0%)1,6).
As an underlying cause of infantile spasms, we experienced the case who had a mutation identified in exon 9 of SLC2A1. Ketogenic diet relieved her of infantile spasms. In previous report, a patient with infantile spasms who did not respond to several antiepileptic drugs diagnosed as GLUT-1 deficiency. He was treated with ketogenic diet and later the seizure stopped11). These observations suggest that earlier detection of GLUT-1 in infantile spasms protects patients from disease progression. When infantile spasms are diagnosed, metabolic screening, including CSF examination, is needed for earlier detection of GLUT-1.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


