


Introduction
Somatic interstitial deletions at the long arm of chromosome 5 [del(5q)] are common in patients with the 5q- syndrome. The 5q- syndrome is a myelodysplastic syndrome characterized by marked preponderance in females, refractory macrocytic anemia, normal or high platelet count, hypolobulated megakaryocyte count, and modest leukopenia1,2,3).
However, constitutional interstitial deletions at 5q are uncommon and deleted segments vary in region and size, which makes it difficult to define a typical syndrome. To our knowledge, only 2 cases with interstitial deletion of the band 5q33.3q35.1 have been reported until date. These cases showed common phenotypic features, including mental retardation and developmental delay4,5).
In Korea, only 1 patient with constitutional interstitial deletion of 5q has been reported6). There are no reports that described severe mental retardation as a result of the interstitial deletion of 5q33.3q35.1. Here, we report, for the first time in Korea, the case of an 11-year-old boy who carries the 46,XY,del(5)(q33.3q35.1) deletion as the probable cause of his mental retardation accompanied with facial dysmorphisms, developmental delay, and epilepsy.
Case report
1. Subject
An 11-year-old boy was referred to the pediatric neurology clinic in Konyang University Hospital for evaluation of severe mental retardation, developmental delay, and seizures.
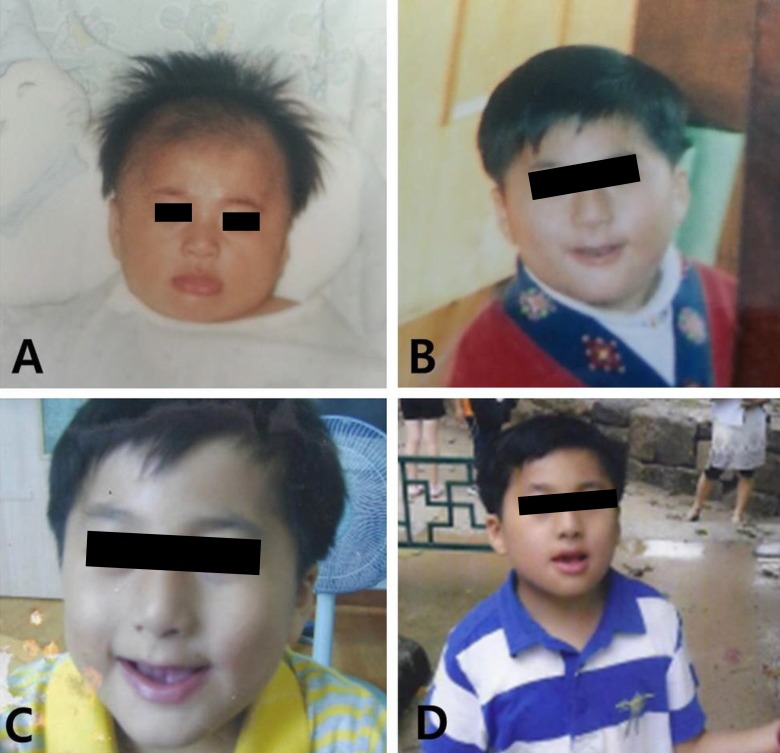
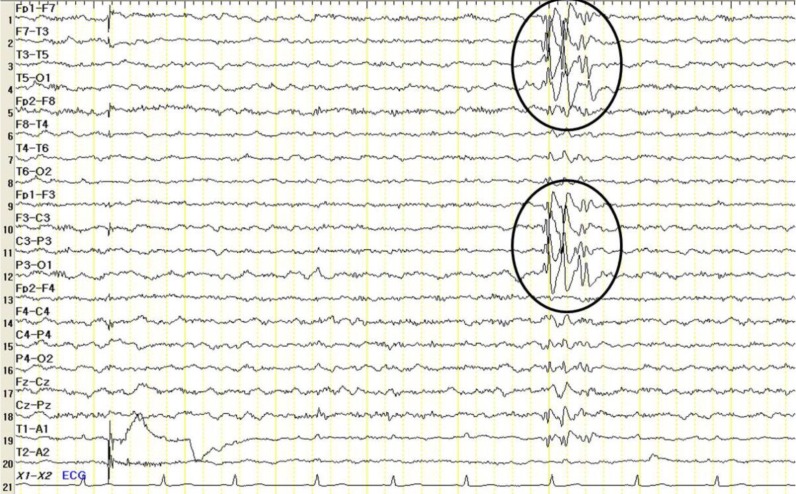
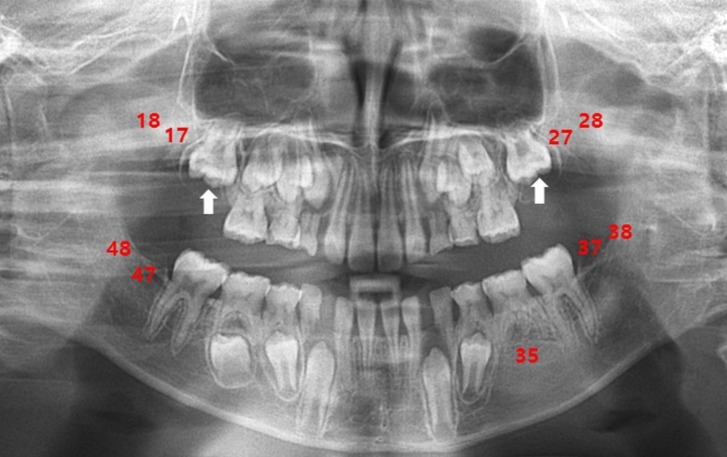
At the time of referral, although his movements were not impaired, he showed overactivity and attention-deficit nature. On physical examination, his weight was 23 kg (less than 3th percentile), height was 120.4 cm (less than 3th percentile), and occipitofrontal circumference (OFC) was 48.5 cm (90th to 95th percentile). He had small deep-set eyes with an apparent hypertelorism, a thin upper lip, a thick and everted lower lip, maxillary micrognathia, and short neck (Fig. 1). He was able to express himself using less than 2 words in incomplete sentences, each made of a word, or repetitive monosyllables. He could not understand demands and these speech-language functions were consistent with the age of 13 months. His cognitive development was uneven, as evidenced by his scores on the Korean Wechsler Intelligence Scale for Children III, which was performed at the age of 10. The verbal intelligence quotient (IQ) scores ranged from 36 on arithmetic to 42 on comprehension, and the performance IQ scores ranged from 43 on block design to 47 on coding, with a full-scale IQ of 31. Ultrasonography of the abdomen was normal and the electrocardiogram revealed an intermittently incomplete atrial-ventricular block. The electroencephalogram showed frequent left central and temporal slow waves (Fig. 2). There were dental anomalies with hypodontia, including all second permanent molars and left mandibular second permanent premolar, and agenesis of all wisdom teeth in a panoramic radiograph (Fig. 3). In addition, delayed eruption of maxillary first permanent molars and poor development of maxillary bones were noticed. His bone age was consistent with his chronological age. Chromosomal microarray (CMA) analysis was performed because of facial dysmorphisms, severe mental retardation, developmental delay, and seizure occurrence.
His medical histories are as follows. He was the third child of healthy, nonconsanguineous parents, a 34-year-old father and a 31-year-old mother at the time of his birth. His mother did not take any medications during the pregnancy and no prenatal growth retardation was observed. There was no known inherited disorder in the family. He was born at a gestational age of 42+0 weeks by vaginal delivery, with a birth weight of 2.9 kg (10th to 25th percentile) and had no perinatal problems. His length was 48 cm (10th to 25th percentile) and his OFC was 32 cm (10th to 25th percentile). His Apgar score was 7 at 1 minute and 8 at 5 minutes. After birth, he was hospitalized because of poor oral intake and fever. He showed subsequent improvement and was discharged without any problems within the next few days. At the time, the hospital records described that he showed facial dysmorphisms such as small eyes with hypertelorism, open mouth, and pectus excavatum. However, he had no symptoms of muscular hypotonia. Excluding chromosomal analysis, no other abnormalities were identified from the laboratory findings. We reviewed his standard chromosomal analysis of peripheral blood lymphocytes, G-banded karyotyping, based on the medical records from the neonatal period. Our analysis revealed that an add(5) was present in all analyzed cells, specifically the 46,XY,add(5)(q31) variant (Fig. 4). However, although a deletion of chromosome 5 at 5q33.3q.35.1 was suspected, unfortunately, it could not be confirmed at that time because of relatively low resolution of G-banded karyotyping. Ultrasonography of the brain revealed the presence of small bilateral cysts in the choroid plexus. A small-sized patent ductus arteriosus was detected in the echocardiogram; however, it was found to have closed spontaneously during follow-up visits. Nuss operation for pectus excavatum was performed at 4 years of age because he intermittently suffered from respiratory discomfort. He also had a follow-up visit for subclinical hypothyroidism.
At 6 months of age, a gradual delay in global development was noticed. For example, he could sit up by the age of 10 months and walk at 3 years. Until 1 year of age, he showed no response in brain stem audiometry. Speech development was also delayed; he started uttering single words only when he turned 4 years old. His first epileptic seizure occurred with fever when he was 3 years old and was first erroneously interpreted as febrile convulsion. Later, the recurrent nature of these seizures became apparent until the age of 8. After treatment with antiepileptic medications, the patient reached a seizure-free state. Magnetic resonance imaging at 8 years of age showed unremarkable findings.
2. Cytogenetic studies
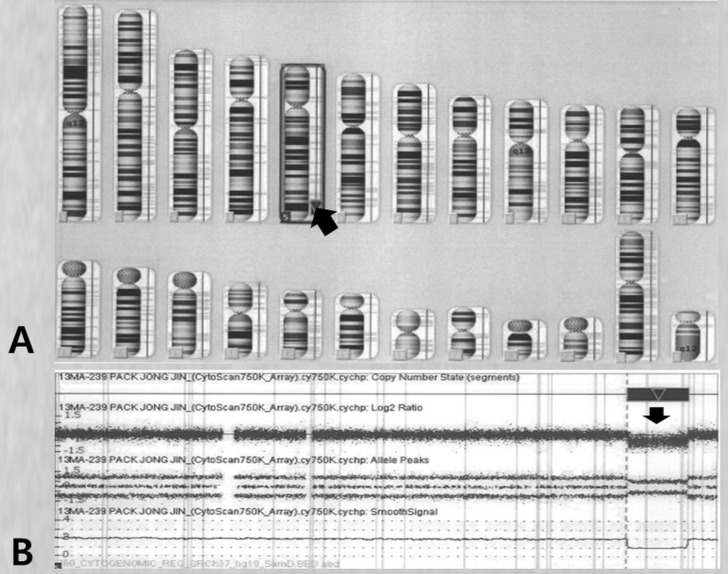
The parents provided a formal informed consent that permitted the use of peripheral blood sampling and genetic testing. We performed high-resolution CMA analysis to specify the breakpoint. The genomic DNA was analyzed using a Cytoscan 750K array and the chromosome analysis suite software package (Affymetrix, Santa Clara, CA, US) using genome build Hg19. The results showed a 16-Mb deletion of 5q33.3q35.11 and an array aberration with 5q33.3q35.1(156,409,412-172,584,708)x1 (Fig. 5). His parents had apparently normal chromosomes. Consequently, the patient was diagnosed as having a de novo deletion in the 5q33.3q35.1 chromosome.
Discussion
The 5q- syndrome is the most well-known disease that develops as a result of somatic interstitial deletions of the long arm of chromosome 5; it was first reported by Van den Berghe et al.2). Five patients were reported to have macrocytic anemia, normal-to-elevated platelet count, dyserythropoiesis, and hypolobulated megakaryocytes. Although deletions in 5q vary among patients diagnosed with the 5q- syndrome, the most frequent deletion is del(5)(q13q33). In nearly all cases studied, the critical deleted region included 5q311,3).
In contrast, constitutional interstitial deletions of 5q are rare and karyotype-phenotype correlations are not well defined in the literature. A small number of 5q deletion cases was reported in association with mental retardation and facial dysmorphism, like in our case. Lee et al.7) documented patients carrying the 5q22.3 deletion. These cases included clinical features of brachycephaly, a high forehead, hypertelorism with prominent eyes, low-set ears, clenched hands, clubfeet, prominent coccyx with hair, ambiguous genitalia, inguinal hernia, cardiac defect, and severe failure to thrive. In addition, a case of interstitial deletion of the long arm of chromosome 5q13q22 was also described in a girl who presented with mental retardation, severe hypotonia, facial dysmorphisms, and peculiar dermatoglyphics8). Alternatively, some of the 5q deletions were known to be related to specific diseases. Several cases were found to have deletions at 5q13q15 or 5q15q22. These deletions were found to be relevant in Gardner syndrome with mental retardation9).
The patient described here had an interstitial deletion in the chromosome band 5q33.3q35.1. Two previously reported cases had involved phenotypic features similar to this case. The first case was of a 4-year-old girl who presented with mild mental retardation, minor facial anomalies, and seizures4). The second case was of a 6-year-old girl who showed overall developmental delay, mild mental retardation, and severe overactivity5). Both cases, including ours, presented with mental retardation as a common feature. We suspect that the observed mental retardation is related to the gamma-aminobutyric acid (GABA) A receptor based on a previous report that the gene for the β 2 subunit of the GABA A receptor is mapped on chromosome 5q3410).
Dental anomalies, one of the major described phenotypes, were unique to our patient, which differentiated him from other cases. The patient presented hypodontia, delayed eruption, and poor development of the maxillary bones. These phenotypes are rarely observed in patients with a deletion of 5q. In particular, hypodontia is genetic in origin and is the term most commonly used to describe conditions of missing teeth from 1 to 6, except for the wisdom teeth11).
Molecular genetics have established the importance of the muscle-specific homeobox genes such as MSX1 and MSX2 in dental development. The MSX1 and MSX2 genes localize to the chromosome 4 and 5, respectively. More specifically, the MSX2 gene is located on the chromosome 5q34-3512). We speculate that the observed dental anomalies and hypodontia in this patient are associated with a deletion of 5q. However, further genomic evaluation is required to identify a clear correlation between dental anomalies and a deletion of 5q. This is primarily because several reports documented that dental anomalies could be regarded as a multifactorial condition and could include a polygenic mode of inheritance with epistatic genes along with environmental factors12).
A couple of genes have been mapped within the chromosomal region of 5q33.3q35.1. One of these genes is the human cardiac-specific homeobox gene (hCsx), which is expressed in the cardiac tissue and may produce several phenotypic abnormalities such as high incidence of congenital heart disease13). Another gene located at chromosome 5q34 is the forkhead family of winged helix transcriptional regulators (Fkh10). These genes are relevant for inner ear development during the embryonic period14). Although our patient showed no response in brain stem audiometry until 1 year of age, there is no previous evidence with respect to the involvement of cardiac tissue or auditory impairment in patients described previously with a deletion of 5q33.3q35.1.
Standard G-banded karyotyping for this patient during the neonatal period offered results different from those obtained for the CMA performed at the age of 10. This is considered a consequence of the limitation of the G-banded karyotyping method; in particular, because of the difficulty to detect small imbalances of a chromosome due to the low effective resolution. Currently, a new consensus has emerged that recommends the use of CMA primarily as a clinical diagnostic test for individuals with developmental disabilities or congenital anomalies15). Hence, we suggest performing CMA on patients with congenital anomalous features in whom abnormalities could not be detected by standard G-banded karyotyping.
This report provides new elements to draw a rough phenotypic map of the long arm of chromosome 5 (5q) and to correlate several phenotypes to specific chromosomal regions, especially to features such as facial dysmorphisms with dental anomalies, mental retardation, developmental delay, and epilepsy. Although interstitial deletion of 5q33.3q35.1 is a rare condition, it is important to accurately diagnose mental retardation to ensure proper genetic counseling and to guide patients as part of long-term management.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


