


Introduction
Paroxysmal kinesigenic dyskinesia (PKD) is an episodic movement disorder characterized by sudden attacks of involuntary movements, such as dystonia, choreoathetosis, and ballism. Most patients with PKD are neurologically normal between the attacks and remain conscious throughout the attacks. The attacks of abnormal movements are triggered by sudden voluntary movements and last less than one minute1,2).
PKD has been associated with mutations in the gene encoding proline-rich transmembrane protein-2 (PRRT2), located on chromosome 16 (16p12.1-q12.1), although the precise role of PRRT2 in PKD remains unclear3). PKD can occur independently of or concurrently with benign infantile convulsions (BIC), a condition characterized by nonfebrile convulsions with onset between 3 and 12 months of age and favorable outcomes, including normal psychomotor development4). The diagnosis of PKD is based on clinical features and the correct diagnosis has implications for treatment and prognosis. However, PKD is often misdiagnosed clinically as epilepsy or psychogenic illness. Recent genetic discoveries will increase the likelihood of a correct diagnosis and contribute to elucidating the pathophysiology of this disease.
Centrotemporal spikes (CTS) are diagnostic marker of benign childhood epilepsy with CTS (BECTS) and it is only a diagnostic marker in patients showing characteristic clinical seizure. CTS are not specific to BECTS as they occur in 2%вҖ“3% of normal school-aged children, of whom <10% develop seizures5). Moreover, CTS are also associated with other diseases6).
This study describes a 10-year-old girl presenting with PKD, BIC and PRRT2 mutation, as well as CTS discharges on electroencephalogram (EEG).
Case report
A 10-year-old girl presented with a 3-year history of short duration dyskinesia after sudden movements. She was born via normal vaginal delivery at 38 weeks and 6 days of gestation, with a birth weight of 3,400 g. Her perinatal history was unremarkable, and she developed normally. At age 6 months, she developed a sudden onset of staring, which progressed to generalized hypotonic seizures that lasted for 3 minutes. Her growth and developmental milestones were normal. EEG, neurologic examination and brain magnetic resonance imaging (MRI) were normal. At age 10 months, after experiencing several afebrile seizures, she was started on treatment with an antiepileptic drug (AED) for about 2 years. Her seizures were well controlled on the AED.
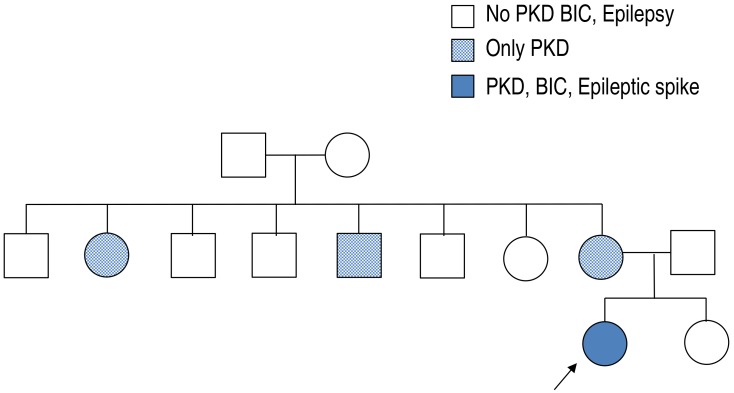
There was no family history of epilepsy or febrile seizures. However, while of school age, her mother and her mother's sister and brother had similar dyskinesia after sudden movements (Fig. 1). Their development was normal with no neurologic problems and no febrile seizures or epilepsy since birth. They no longer have these symptoms.
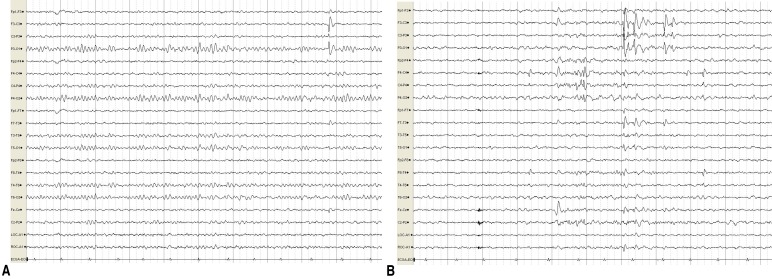
At 7 years of age, she presented with abnormal paroxysmal movements occurring 5вҖ“6 times a day. She staggered while laughing and suddenly collapsed in the street while walking. These symptoms continued to occur. The results of neurologic examinations and brain MRI were normal. EEG revealed bilateral CTS, and there were no changes in EEG during movements (Fig. 2).
After obtaining informed consent from her parents, a blood peripheral sample was obtained from this patient, its DNA was extracted and her PRRT2 gene was sequenced. A heterozygous frameshift mutation, .649dup, was detected, resulting in p.Arg217Profs*8 (Fig. 3).
Two to 3 days after oxcabarzepine therapy (3 mg/kg/day), her paroxysmal movements were well controlled. She continued oxcabarzepine thereafter and continued to develop normally, with no further seizure and no attacks of PKD.
Discussion
PKD is a paroxysmal movement disorder characterized by recurrent, brief attacks of abnormal involuntary movements induced by sudden voluntary movements. The episodes usually present with dystonia, chorea, athetosis, ballism, or a combination of these symptoms. The attacks usually last from a few seconds to a few minutes. During these attacks, there is absolutely no loss or alteration of consciousness. The prognosis of PKD is favorable and patients usually show excellent response to treatment. PKD can be classified as primary or secondary according to its etiology. Most cases of secondary PKD are caused by multiple sclerosis, head injury, metabolic derangements, or cerebral perfusion insufficiency. Primary PKD can be classified as idiopathic or familial. Familial PKD is more common, with the condition usually inherited in an autosomal dominant manner7).
In 2011вҖ“2012, mutations in the PRRT2 gene were found to be responsible for PKD and infantile convulsion with choreoathetosis syndrome7). The molecular function of the PRRT2 protein has not been fully defined. PRRT2 is likely expressed in the brain and spinal cord during the embryonic and postnatal stages of development3,5,7). PRRT2 is thought to interact with synaptosomal-associated protein 25 (SNAP25), a member of the family of soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins, which fuse synaptic vesicles to the presynaptic plasma membrane and release neurotransmitters. Defects in synaptic functions are predicted to be associated with brain disorders. Moreover, SNAP25 is thought to modulate the kinetics of voltage-gated Ca2+ channels. In patients with PRRT2 mutations, the interaction between PRRT2 and SNAP25 might be affected, resulting in an alteration of Cav2.1 properties. These data thus suggest that PKD, benign familial infantile seizures, infantile convulsions with choreoathetosis and hemiplegic migraine could result from dysfunction of a protein involved in synaptic regulation, with ensuing neuronal hyperexcitability8).
Previous reports have indicated that mutations in the PRRT2 gene may cause PKD, infantile convulsions, familial hemiplegic migraine9), episodic ataxia10) and febrile seizures11). Also, recent study showed that a link between BECTS, BIS, PKD or exercise-induced dystonia and PRRT2 mutations detected by array comparative genomic hybridization and quantitative polymerase chain reaction12,13). A subset of genomic alterations detected in BECTS contains candidate or known epilepsy genes including PRRT2, some of which are likely to influence the emergence, the presentation, or the comorbidity of BECTS12).
BECTS is considered the most common type of childhood epileptic syndrome, present in 10%вҖ“20% of pediatric patients with epilepsy. It is classified among the idiopathic localization-related epilepsies. Characteristic EEGs show high voltage spikes or spike and waves in the centrotemporal region, which may shift from side to side on a normal background. Neuro-imaging is normal. CTS are diagnostic markers of BECTS only in a suggestive clinical presentation. CTS are not specific to BECTS, however, as they occur in 2%вҖ“3% of normal school-aged children, of whom <10% develop seizures. They are common among relatives of children with BECTS and may also occur in various organic brain diseases, with or without seizures, such as cerebral tumors, Rett syndrome, fragile X syndrome and focal cortical dysplasia. They may incidentally be found in nonepileptic children with various symptoms, such as headache, speech and behavioral and learning difficulties6). PRRT2 heterozygous mutations cause a wide variety of variably associated paroxysmal cerebral disorders: BIC, PKD, and/or hemiplegic migraine. So we can infer that the mutation in PRRT2 contributed to the development of BIC, paroxysmal disease, and the diseases showing CTS on EEG or BECTS.
Further functional study of PRRT2 mutations should assist in elucidating the pathophysiology of various paroxysmal disorders including epilepsy, movement disorders, and disease associated with CTS on EEG, as well as other neurologic disorders.
PKD is often misdiagnosed clinically as psychogenic illness or epilepsy, especially in patients with CTS on EEG, which may cause great distress to patients. A correct diagnosis and subsequent treatment with low doses of anticonvulsants can eliminate the clinical manifestations of PKD and improve patient quality of life in patients.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


