


Introduction
Anti-N-methyl D-aspartate receptor (anti-NMDAR) encephalitis, first described in young women as a paraneoplastic disorder in association with ovarian teratoma in 20071), is recently characterized as a severe neurological autoimmune disease with a progressive clinical course. Its diagnosis is based on the clinical course of the condition and specific neurological features. It is mainly characterized by psychiatric symptoms with behavioral features or cognitive disturbances, followed by seizures, altered consciousness, movement disorders such as orofacial dyskinesia and dysautonomia. Positive detection of anti-NMDAR antibodies in the serum and cerebrospinal fluid (CSF) is essential for its diagnosis. Prompt diagnosis and treatment are important for full recovery.
Anti-NMDAR encephalitis in children is increasingly recognized as one of the commonly identified causes of encephalitis over the last 5 years2,3), and has different symptom presentation and tumor association compared to that in adults2,3,4). Although its natural history for pediatric patients is not well defined, seizures, behavioral changes, and movement disorders are prominent. Occurrence of tumor is low in pediatric patients with anti-NMDAR encephalitis3,5).
Here, we present a case of a young child with anti-NMDAR encephalitis that was not associated with tumor. Initial symptom of epilepsia partialis continua at onset was observed, followed by orofacial-limb dyskinesias and cognitive impairment. The patient showed significant improvement in symptoms after administration of rituximab.
Case report
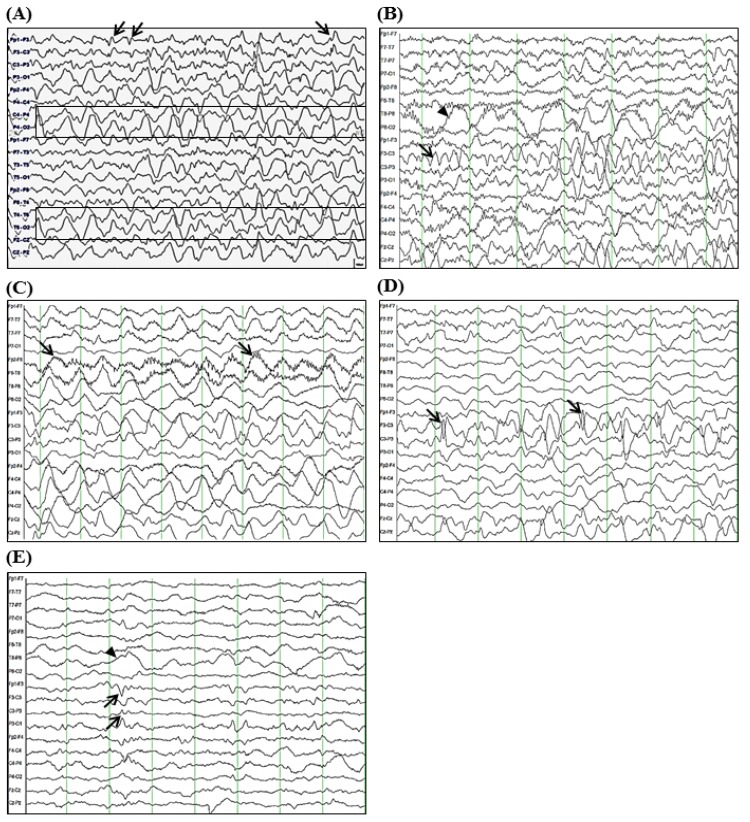
A 3-year-old girl was admitted to the Department of Pediatrics at Samsung Changwon Hospital. Her major complaint was focal seizure for 15 minutes characterized by tonic-clonic seizure of right arm and leg with lip smacking and unresponsiveness without antecedent febrile illness. She was born by caesarean section at 36 weeks of gestation with a birth weight of 2,600 g. She had mildly delayed motor development. Initial neurologic examination revealed alert mentality without focal neurologic deficits. Although brain magnetic resonance imaging (MRI) and electroencephalogram (EEG) were unremarkable at initial presentation, oxcarbazepine (12.5 mg/kg/day, Trileptal, Novartis Pharma AG, Basel, Switzerland) was administered to her due to recurred seizure for 3 minutes on day 3 after onset. However, on day 5 after onset, her seizure evolved into epilepsia partialis continua without changes in neurological status or fever. She had right-sided focal seizures for 80 minutes that was stopped after loading phenytoin and phenobarbital (20 mg/kg/day, respectively). Repeated interictal EEGs revealed sharp wave discharges on the left frontal areas and 1- to 2-Hz high amplitude delta activities on the right hemisphere (Fig. 1A). CSF was not tested due to low probability of inflammatory or metabolic encephalopathy.
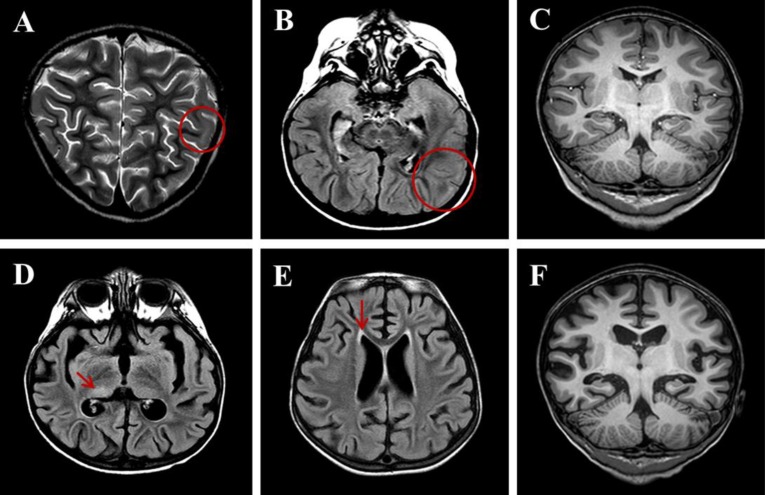
Although her seizures were slowly improved by multiple antiepileptic drugs, including oxcarbazepine, phenytoin, and phenobarbital without alteration of mentality or focal neurologic deficit, she was transferred to the Department of Pediatrics at Asan Medical Center due to repetitive partial seizures and decreased physical activity at 2 weeks after the onset. Follow-up interictal EEGs revealed a 4–6 Hz sharply contoured theta activities on the left fronto-central area and a 1- to 2-Hz high amplitude delta activities on the right parieto-occipital areas (Fig. 1B), whereas repeated brain MRIs identified hyperintense lesion in the subcortical region of the left posterior parietal lobe in T2-weighted and fluid-attenuated inversion recovery (FLAIR) images (Fig. 2A-C). While her seizures were controlled by phenobarbital and oxcarbazepine, she suffered from sleep disturbance. In addition, she developed noticeable oro-lingual-facial dyskinesia, choreoathetoid movements, and hypertension (systolic/diastolic blood pressure 120/83–142/93 mmHg, >99th percentile) during the third week after onset. The dyskinesia did not have an epileptic correlation in EEG monitoring. EEG showed a diffuse delta activity with superimposed rhythmic beta frequency activity (“extreme delta brush” pattern) (Fig. 1C). Infectious etiology was unlikely as she continued to deteriorate with these characteristic symptoms. A potential autoimmune cause, particularly anti-NMDAR encephalitis, was considered at that time. CSF analysis showed 2 white blood cells/µL (3% lymphocytes), 0 red blood cell/µL, total protein of 15 mg/L, glucose of 71 mg/dL, albumin of 4.4 mg/dL, IgG of 2.3 mg/dL, and positive oligoclonal band with IgG index of 2.79. The patient's serum and CSF studies for paraneoplastic encephalitis (antibodies to Hu, Yo, and Ri) and possible known autoimmune etiologies that could cause an encephalopathy, including antinuclear antibody, antithyroid antibodies, and antineutrophil cytoplasmic antibodies, were all unremarkable. The patient's serum and CSF were sent to the Laboratory of Neurotherapeutics at the Department of Neurology, Seoul National University Hospital to screen for anti-NMDAR antibodies using a cell-based indirect immunofluorescence assay using patient's serum and/or CSF6). At 4 weeks after the onset, she was tested positive for the presence of antibodies to NMDA receptor in her serum and CSF. Screening with whole body positron emission tomography, abdomen-pelvis computed tomography for malignant tumor, serological or CSF studies for infectious encephalitis, and inborn errors of metabolism were all unremarkable.
The patient was started with a course of intravenous immunoglobulin (IVIG) therapy (500 mg/kg/day for 4 days). The patient showed no improvement in behavior or level of consciousness. She began to have self-limited episodes of hypertension, tachycardia, hyperthermia, and autonomic instability. Therefore, she was treated with 2 courses of methylprednisolone (30 mg/kg for 5 days) over 2 weeks. After 3 weeks of immunotherapy in conjunction with aggressive physical rehabilitation, her sleep disturbance, dyskinesia, cognitive dysfunction, and dysautonomia were slowly improved, although repeated EEG and brain MRI revealed an ongoing encephalitic process (Figs. 1D, 1E, and 2D-F).
Subsequently, the patient was transferred to Samsung Changwon Hospital for continued intensive behavioral, cognitive, and physical rehabilitation eight weeks after the diagnosis. In addition, to have more favorable outcome, she was treated with weekly intravenous rituximab (a monoclonal anti-CD20 antibody, 375 mg/m2/week, Mabthera, Hoffmann-La Roche AG, Basel, Switzerland-) for a total of 4 doses according to the administration protocol of rituximab. She did not have complications such as serious infection or viral reactivation during the treatment of rituximab. At present, she is no longer taking multiple antiepileptic or antihypertensive drugs. She continues to show gradual motor and cognitive function improvement without relapse. At 12 months follow-up, she showed further improvements. She was able to stand holding onto something, although she required constant help/assistance to organize herself. She is able to follow single stage commands and give some limited verbal responses. Her mood remains relatively stable.
Discussion
Anti-NMDAR encephalitis has been recognized as the most frequent autoimmune encephalitis in children after viral encephalitis or acute demyelinating encephalomyelitis7). As noted in previous case reports, clinical presentation of anti-NMDAR encephalitis in children is often different from that in adults2,3,4). Adult patients usually present with acute behavioral change and psychosis followed by seizures, dyskinesias, memory and language impairment, and autonomic and breathing dysregulation1). However, most children with anti-NMDAR encephalitis have more frequent neurological symptoms, such as seizures and orofacial dyskinesias or choreoathetoid movement of limbs rather than psychiatric symptoms at the disease's onset. Children with anti-NMDAR encephalitis have the rare development of mono-symptomatic illness with less severe autonomic manifestations2,3,4). A recent study showed that seizures at the onset of anti-NMDAR encephalitis were predominant within a group of prepubertal children, suggesting that modification of hormonal activity related to puberty could represent a key factor in the progression of the clinical presentation of anti-NMDAR encephalitis at different ages4). In some cohorts, most pediatric patients presented with partial motor or complex seizures, whereas generalized tonic-clonic seizures or status epilepticus were infrequently presented3,8). Cases of epilepsia partialis continua in pediatric anti-NMDAR encephalitis were rarely reported9,10,11) (Table 1). Our patient also presented with epilepsia partialis continua following repetitive partial seizures. As an uncommon initial symptom in anti-NMDAR encephalitis leads to diagnostic difficulties in the course of the first days following the onset. However, subsequent repetitive orofacial dyskinesias, dystonic or choreoathetoid movements of the limbs, and cognitive deficits were important clinical clues to suspect and diagnose anti-NMDAR encephalitis eventually.
Although laboratory findings of CSF, EEG, and brain MRI are not diagnostic tools for anti-NMDAR encephalitis, some characteristic findings have been suggested by some studies. For our patient, CSF studies revealed no abnormal finding other than an oligoclonal band. CSF analysis in patients with anti-NMDAR encephalitis usually identifies lymphocytic pleocytosis with frequent oligoclonal banding1). However, a recent study reported that the frequency of CSF alteration was lower in children than in adults2). On EEG, our patient showed focal or diffuse delta-theta rhythms on EEG, sometimes with extreme delta brush, a characteristic pattern of anti-NMDAR encephalitis2). Disorganized and diffuse or focal (predominantly frontotemporal) slowing of electrical activity in the delta-theta range, sometimes with rhythmical appearance or epileptic activity on EEG, has been described in children with anti-NMDAR encephalitis similar to that reported in adults3,5). Our patient had variable abnormalities on brain MRI. T2/FLAIR hyperintense lesions on the left parietal lobe at the initial stage and abnormalities on the right thalamus and the bilateral periventricular white matter during the dyskinetic stage of the disease were found. Diffuse cerebral atrophy was revealed on the follow-up image. In most cases, MRI of the brain is usually normal or shows non-specific focal changes at the initial stages of anti-NMDAR encephalitis. Increased signal on T2 or FLAIR MRI sequences could be revealed in basal ganglia, medial temporal lobes, brainstem, or cerebellum1,3). White matter changes and cerebral atrophy have been noted as atypical MRI changes in a few cases of anti-NMDAR encephalitis5).
As noted in previous case reports, most children with anti-NMDAR encephalitis do not have an underlying tumor2,3). Similarly, our patient had no identifiable tumor. Although the mechanisms that initiate this disorder are unknown, it has been suggested that the presence of a tumor that expresses NMDAR likely triggers the immune response. However, the low frequency of paraneoplastic etiology in children with anti-NMDR encephalitis suggests that other immunological triggering mechanisms are associated with postinfectious auto-immune process or an underlying genetic predisposition for disease onset3). Although the triggering mechanism remained unclear in our patient, the absence of a malignancy and good clinical outcome without relapse suggest that a postinfectious auto-immune process is possibly involved.
In vivo and in vitro studies have shown that the structural and functional effects of anti-NMDAR antibodies would result in specific reduction of the levels of synaptic NMDAR by a mechanism of antibody capping, crosslinking, and internalization of the receptors12). In addition, the anti-NMDAR antibodies abrogate NMDAR-mediated currents, potentially altering the mechanisms of synaptic plasticity and enhancing the excitability of the motor cortex13). Overall, these antibody effects coupled with the characteristic clinical syndrome have contributed to the development of treatment strategies by not only removing antibodies from the serum, but also abrogating the inflammatory infiltrates and the synthesis of antibodies within the CNS.
Although there is no uniform systematic treatment approach, treatment of anti-NMDAR encephalitis in childhood is focused on the empirical use of immunosuppressive therapies and supportive care. In initial studies largely involving patients with paraneoplastic disease, response rates to tumor removal and first-line immunotherapies (such as corticosteroids, IVIG and plasmapheresis) were high1). However, it is recognized that tumor-negative patients may not respond to first line immunotherapy. A recent review of anti-NMDAR encephalitis suggested that cyclophosphamide and rituximab should be used as second-line immunotherapies in these patients14). Although long term safety data and formal clinical trials are lacking, rituximab has provided clinical benefits in children with autoimmune encephalitis. The most commonly used regimen of rituximab is 375 mg/m2 weekly for 4 weeks15). Our pediatric case with nonparaneoplastic anti-NMDAR encephalitis was treated with rituximab without apparent side effects. Her outcome was good, although her recovery was slow. Some studies reported that most children with anti-NMDAR encephalitis had remarkable clinical improvement or full recovery2,4). Recent research by Armangue et al.2) reported that 85% of pediatric patients had remarkable improvement or full recovery, although the recovery was achieved 8–12 months after the symptom onset. These results suggest that the production of anti-NMDAR antibodies within the central nervous system as well as systemic production might have contributed to the systemically slow response to immunotherapy and prolonged the duration of disease. Lower severity of symptoms and early treatment of immunotherapy as well as tumor removal are likely to reduce relapses and limit morbidity associated with anti-NMDAR encephalitis4).
In summary, we report the first case of a Korean child with anti-NMDAR encephalitis, initially presenting with focal status epilepticus followed shortly by other characteristic manifestations. Anti-NMDAR encephalitis is an important cause of neuropsychiatric deficits in children, which has to be considered as the differential diagnosis in children with uncontrolled seizures followed by the development of dyskinesias, even if they are young age without evidence of a malignant tumor. Early recognition and aggressive immunosuppressive therapies are essential in order to improve outcomes.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


