


Introduction
Severe aplastic anemia (SAA) is a fatal disease characterized by pancytopenia and hypocellular bone marrow. Bone marrow transplantation is the treatment of choice for young patients who have a human leukocyte antigen (HLA) matched sibling donor. Immunosuppressive treatment (IST) with antithymocyte globulin (ATG) and cyclosporine is an effective therapy for patients who have no suitable donor. ATG exerts its action by depleting clonal suppressor T cells implicated in the pathogenesis of SAA1,2,3). The depletion of suppressor T cells by ATG could change the B-cell activation1,2). Therefore this treatment might increase the risk of autoimmune phenomena. There have been rare reports on the association between ATG treatment for SAA and the development of autoimmune hyperthyroidism4,5,6,7).
Because horse ATG (hATG) is no longer available for the treatment of SAA in most countries, rabbit ATG (rATG) is the only preparation currently available8). Moreover, as rATG has more potent and prolonged suppression of T cells than hATG8), more frequent encounters of autoimmune hyperthyroidism can be anticipated. We report the rare case of Graves disease following rATG treatment for SAA in a Korean child.
Case report
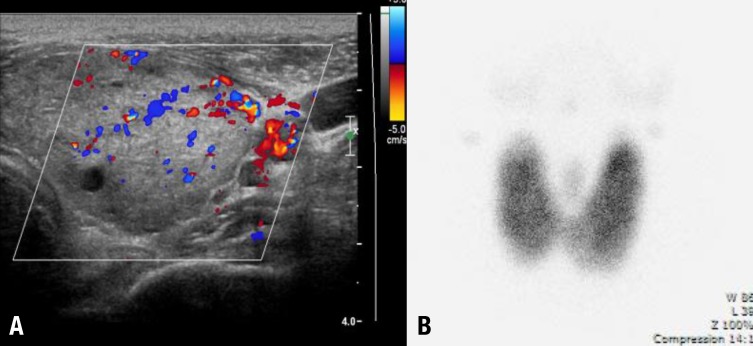
A 12-year-old boy visited Chonnam National University Hwasun Hospital because of petechiae, easy bruising and knee joint pain in December 2007. There was no relevant past medical history. A complete blood count showed white blood cell 1,600/µL, hemoglobin 11.7 g/dL, platelets 4,000/µL, absolute neutrophil count 940/µL, and corrected reticulocyte count 0.89%. A bone marrow biopsy showed a hypocellular fatty marrow (cellularity <10%). Chromosome breakage test was negative. Cytogenetics was normal. Flow cytometry analyses for paroxysmal nocturnal hemoglobinuria were negative. The patient was diagnosed as a SAA. As he had no HLA-matched sibling, he was treated with IST using rATG (Thymoglobuline, Genzyme, Cambridge, MA, USA; 3.5 mg/kg/day for 5 days) and cyclosporine (5 mg/kg/day) and methylprednisolone (2 mg/kg/day for 5 days, then tapered over 30 days). While being treated with IST, he developed peripheral neuropathy with pain and tingling sensation in both extremities, requiring a sympathetic nerve block, which relieved symptoms. Five months after the IST, he became transfusion independent. Twenty-three months after IST with rATG, he complained of mild hand tremors, sweating, weight loss, palpitations and goiter. Subsequent thyroid function tests showed the hyperthyroidism (free thyroxine, 3.42 ng/dL; TSH, <0.01 nIU/mL; triiodothyronine, 3.99 ng/mL; thyroxine, 17.3 µg/dL). Autoantibody results were as follows: antimicrosome antibody (+), TSH-binding inhibitory immunoglobulin (+), antithyroglobulin antibody (-), and antinuclear antibody (-). Thyroid ultrasonography showed diffuse thyroid enlargement with multiple benign nodules (Fig. 1A). A Tc-99m pertechnetate thyroid scan showed enlarged thyroid gland with diffuse uptake (Fig. 1B). Thus, he was diagnosed with Graves disease. He was treated with methimazole and the symptoms improved. The patient has been disease free 39 months after IST and 9 months after methimazole treatment. Currently, his blood counts and thyroid function tests are normal, and he has no discomfort from Graves disease.
Discussion
Graves disease is an autoimmune disorder characterized by hyperthyroidism, diffuse goiter, ophthalmopathy, and occasionally a dermopathy referred to as pretibial or localized myxedema9). In immune pathogenesis of Graves disease, B and T cells have a major role in the production of TSH receptor antibodies (TSHR-Ab) that are responsible for the thyroid stimulation and growth. T cells are present in the immune repertoire of patients with Graves disease. These activated T cells in turn increase the secretion of TSHR-Ab from B cells. The hyperthyroidism and goiter of Graves disease are caused by stimulation of the thyroid by TSHR-Ab9). It has been suggested that a genetic clonal lack of suppressor T cells may be responsible for the unregulated production of TSHR-Ab in in vitro studies10).
The pathogenesis of aplastic anemia is thought to be T-cell-mediated destruction of hematopoietic progenitors and stem cells. A variety of in vitro and in vivo research supported the implication of immune effector cells and cytokines, such as interferon-γ or tumor necrosis factor-α1). The mechanism action of ATG therapy for aplastic anemia is likely complex and multifactorial, but its main mechanism is thought to have immunosuppressive effect by depleting peripheral T cells from the circulation through complement-dependent lysis or activation associated apoptosis11). ATG is manufactured through the immunization of horses or rabbits with thymocytes or T-cell lines, followed by the purification of the IgG fraction of the sera from these animals. Between hATG and rATG preparations, there are differences in pharmacologic properties, antibody specificity and binding capacity. Thus, they are dissimilar in inducing activation of T cells, as exemplified in the case of expansion of regulatory T cells11). Also, gene expression profiling was markedly different between cells treated with both preparations11). As a consequence, hATG and rATG would provide different mechanisms of action through multiple modes of immunomodulation. Despite the differences mentioned above, hATG and rATG have generally been used interchangeably in the clinic. However, in a recent randomized, prospective trial, rATG plus cyclosporine compared unfavorably with hATG plus cyclosporine when used as a first-line treatment. These results were unexpected, given that rATG is more immunosuppressive than hATG12). The use of rATG led to a more profound depletion of CD4+ T cells, whereas both preparations resulted in a similar depletion of CD8+ T cells8).
The depletion of suppressor T cells by ATG was thought to have a role in the development of many autoimmune disorders3). Interferon-γ is vital for cell-mediated immunity, inhibition of inappropriate B-cell proliferation and antibody production. It is secreted by a subset of suppressor T cells to inhibit Th2-cells which in turn activate B cells13). Thus, depletion of suppressor T cells and resultant decreased production of interferon-γ by ATG could also allow B-cell activation, which increases the risk of autoimmune disorder. This was supported by studies showing that interferon-γ down-regulates TSHR-Ab production14). It also attenuated TSH receptor (the TSHR-Ab target) synthesis in Graves disease15). Thus, impaired interferon-γ production can cause derestricted TSHR-Ab production and increased TSH receptor production, resulting in immune hyperthyroidism.
There are a few reports of Graves disease after treatment with ATG for SAA4,5,6,7). Three cases have been described occurring 26, 42, and 106 months after treatment with antilymphocyte globulin or ATG in patients aged 25, 30, and 35 years, respectively4). A 62-year-old man developed Graves disease and fibrosing alveolitis 15 days after the administration of ATG for SAA5). The first case in children was described 6 years after ATG in a 10-year-old boy7). All the reported cases in the literature were treated with horse preparations.
This case represents the first case of possible implication of rATG in the pathogenesis of autoimmune hyperthyroidism. Recently, hATG is no longer available for the treatment of SAA in Europe, Japan, Latin America, and Korea. Moreover, rATG has more potent and prolonged suppression of T cells than hATG8), and the development of autoimmune hyperthyroidism may be encountered more frequently. However, the incidence and exact mechanism underlying the association should be elucidated. Therefore, thyroid function tests should be incorporated in the routine follow-up of SAA following rATG treatment.
 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


