


Introduction
Hepatomegaly and elevated liver enzyme levels in type 1 diabetes mellitus (DM) patients can be caused by hepatic glycogenosis, viral hepatitis, autoimmune hepatitis, or metabolic liver diseases. Mauriac syndrome is a rare complication of type 1 DM that is characterized by hepatomegaly due to hepatic glycogenosis, growth retardation, delayed development of puberty, and Cushingoid features. It is caused by abnormal hormone secretion and metabolic abnormalities because of inappropriate glycemic control1). If the insulin dosage is insufficient to control hyperglycemia, it can stimulate hepatic glycogenosis and fatty acid synthesis, resulting in inhibition of glycogenolysis and hepatomegaly. Hepatic glycogenosis in type 1 DM patients is a reversible complication by strict blood glucose level control, which is different from nonalcoholic steatohepatitis (NASH) in obese type 2 DM. Thus far, eight cases of type 1 DM accompanied by hepatic glycogenosis and three cases of Mauriac syndrome have been reported in Korea2,3,4). We report a case of hepatic glycogenosis mimicking Mauriac syndrome in a patient with poorly controlled type 1 DM who showed hepatomegaly, Cushingoid face, mild growth retardation, and delayed menarche.
Case report
A 14-year-old girl was admitted to The Catholic University of Korea, Seoul St. Mary's Hospital because of right upper-quadrant abdominal pain and distension. The patient was diagnosed with type 1 DM at the age of 9 years. She had been receiving treatment according to a mixed-split insulin regimen. Total daily insulin dosage was 12 IU (0.26 IU/kg/day). The insulin dosage was insufficient to control hyperglycemia because of recurrent hypoglycemia. She had previously been admitted seven times because of diabetic ketoacidosis in the previous 2 years.
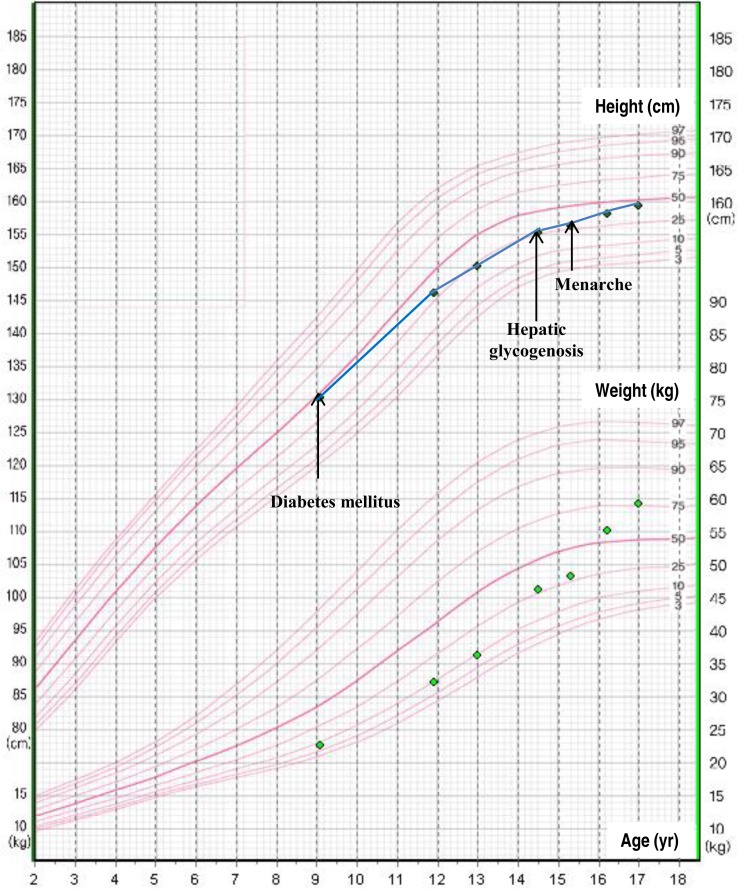
At the time of admission, the patient had clear consciousness, with the following vital signs: blood pressure, 90/60 mmHg; pulse, 118/min; and body temperature, 36.5℃. The height of the patient was 155 cm, and her weight was 46 kg, which were in the 25th and 50th percentiles, respectively. Growth velocity decreased to 3.3 cm/yr in the past 2 years (Fig. 1). With respect to pubertal development, the patient presented with Tanner stage IV breast and Tanner stage III pubic hair; however, the patient did not experienced menarche. Bone age was delayed by 2 years compared to her chronological age. Upon physical examination, the patient showed Cushingoid face (Fig. 2). Liver was palpated at 11 cm below the right costal margin with mild tenderness.
Blood test results revealed the following values: white blood cells, 4,840/mm3 (47.5% neutrophils and 42.6% lymphocytes); hemoglobin, 13.4 g/dL; platelets, 218,000/mm3; aspartate aminotransferase (AST), 1,408 IU/L; and alanine aminotransferase (ALT), 459 IU/L; gamma glutamyltransferase, 430 IU/L; and amylase, 115 IU/L. The glycosylated hemoglobin (HbA1c) level was 14.3%, and ketone was not detected in urine. To rule out viral hepatitis, we performed serologic tests for hepatitis A, B, and C; Epstein-Barr virus; and cytomegalovirus infections, and obtained negative results. Concentrations of alpha-1-antitrypsin and ceruloplasmin were normal. We excluded alpha-1-antitrypsin deficiency and Wilson disease. Furthermore, the results of autoimmune hepatitis, including antinuclear antibody, antismooth muscle antibody, and antiliver kidney microsome antibody, were all negative. Thyroid function was normal and the following hormone levels were presented: insulin-like growth factor-1 (IGF-1), 203 ng/mL; luteinizing hormone (LH), 4.47 mIU/mL; follicle-stimulating hormone (FSH), 4.77 mIU/mL; and estradiol (E2), 35.44 pg/mL. The plasma cortisol level was 5.73 µg/dL and the adrenocorticotropic hormone level was 17.14 pg/mL at 8:00 AM.
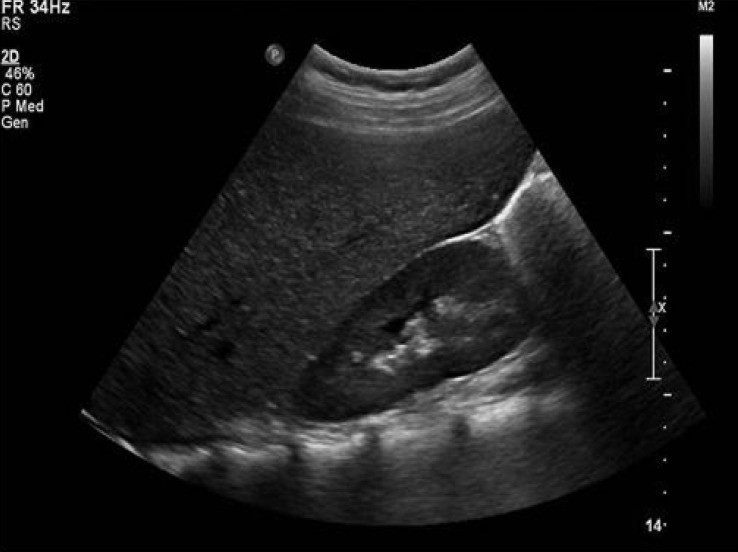
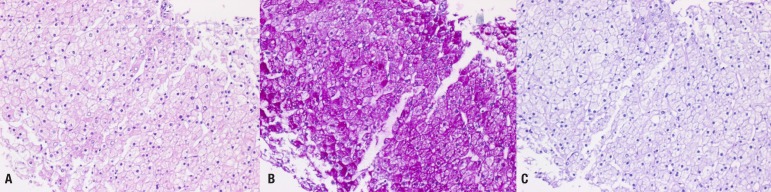
Abdominal ultrasound revealed hepatomegaly and an increase in hepatic parenchymal echogenicity (Fig. 3). Through a liver biopsy, we found ballooning degeneration with nuclear vacuolization of hepatocytes (Fig. 4A). Hepatocytes were strongly positive in a Periodic acid-Schiff (PAS) stain (Fig. 4B), and such positive finding disappeared with diastase (D-PAS) (Fig. 4C).
Continuous glucose monitoring showed hyperglycemia after meals and hypoglycemia before meals. We changed the insulin treatment regimen to an insulin pump for controlling hyperglycemia. The insulin dosage was gradually increased over 3 months; thus, the total daily insulin dosage became 36 IU (0.65 IU/kg/day). To prevent hypoglycemia, we prescribed 60 g (1.3 g/kg) of uncooked cornstarch between meals and bedtime. After implementing strict glycemic control, hepatomegaly was improved and elevated transaminase levels returned to normal ranges. The HbA1c level decreased to 10.5% after 3 months. Moreover, the patient manifested menarche at the age of 15.3 years and a regular menstruation cycle thereafter.
Discussion
Hepatic glycogenosis in type 1 DM is a key mechanism of Mauriac syndrome and is a rare diabetic complication first reported by Mauriac in 19305). Hepatic glycogenosis can be caused by either a persistent hyperglycemia due to insulin deficiency or conversion of excess glucose administered to control hypoglycemia due to excessive insulin administration. With regard to the latter mechanism, the injected glucose penetrates hepatocytes through passive diffusion and is converted to glucose-6-phosphate. The synthesized product is then converted to glycogen by glycogen synthase6,7,8). Furthermore, the secretion of the antagonistic hormone cortisol increases in type 1 DM patients with poor glycemic control. Such excess cortisol inhibits hepatic glycogenolysis and increases fatty acid synthesis in the liver, causing hepatomegaly. Moreover, hypercortisolism can result in Cushingoid features and delay in bone maturation, which in turn leads to delayed growth and puberty1).
Growth retardation and delayed puberty with hepatic glycogenosis in uncontrolled type 1 DM patients are distinct characteristics of Mauriac syndrome. In patients with Mauriac syndrome, circulating IGF-1 levels are normal or reduced despite normal hypothalamic-pituitary functions. Patients with Mauriac syndrome also exhibit resistance to growth hormones, which is caused by a decrease in IGF-1 bioactivity, IGF-1 receptor defects, and circulating IGF-1 inhibitors9,10,11,12). Delayed puberty can manifest as a result of insulin deficiency, hyperglycemia itself, or decreased LH and FSH secretion caused by the gonadotropin-releasing hormone due to stress hormone secretion after poor glycemic control. Insulin administered to the subcutaneous tissue bypasses the hepatic first-pass metabolism and is absorbed directly via systemic circulation. Then, the insulin binds with the receptor at the ovary to excessively stimulate the ovary, thereby increasing androgen secretion. If peripheral insulin resistance increases because of hyperglycemia, polycystic ovary syndrome can occur, causing ovarian failure and amenorrhea13,14).
Although growth retardation and delayed puberty were not prominent in our patient, the patient exhibited very similar clinical manifestations of Mauriac syndrome. The reason that the signs of growth retardation and delayed puberty were mild in our patient was probably because hepatic glycogenosis occurred in the late stage of pubertal development, thereby only minimally affecting the patient's growth and puberty. As we did not measure 24-hour urinary free cortisol levels and the 8:00 AM. cortisol levels were normal, we could not confirm hypercortisolism. If the symptoms of Mauriac syndrome recur, a detailed evaluation of hypercortisolism will be necessary.
Treatments of hepatic glycogenosis in type 1 DM include strict glycemic control through appropriate diet and insulin dosage. Although regular diet and sufficient insulin dosage are important in controlling hyperglycemia, determining the appropriate insulin dosage is difficult when hyperglycemia is accompanied by frequent hypoglycemia, as in the case of our patient. As glycogenolysis does not properly occur even with hypoglycemia in the patient with hepatic glycogenosis, additional glucose supply is necessary. However, the glycemic effects of conventional snacks only manifest 2 hours after consumption. Moreover, as the glycemic effect inevitably decreases with time, hypoglycemia can recur. Consequently, consuming uncooked cornstarch to induce delayed glycemic effects facilitates the prevention of hypoglycemia15).
Hepatic glycogenosis does not occur in all patients with type 1 DM who have poor glycemic control. Although the risk factors of hepatic glycogenosis are not yet known, we postulate that wide fluctuations of blood glucose levels and a long period of poor glycemic control increase the risk of hepatic glycogenosis. However, hepatic glycogenosis in type 1 DM is a reversible complication that is different from NASH, which is often presented in obese type 2 DM patients. NASH is a progressive disease that can lead to liver fibrosis and cirrhosis. Therefore, appropriate glycemic control is important to the treatment and prevention of hepatic glycogenosis in type 1 DM.

 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


