

Introduction
Mucopolysaccharidosis type II (MPS II, Hunter syndrome; MIM 309900) is an X-linked degenerative disorder caused by the deficiency of the lysosomal enzyme iduronate-2-sulfatase (IDS)1). The predominant clinical characteristics of MPS II include coarse facial features, stiff joints, hepatosplenomegaly, cardiovascular and respiratory disorders, developmental delay, and mental retardation2). MPS II manifests with a wide phenotypic spectrum and cases of this disorder can be classified into severe and attenuated types. The severe type is characterized by an onset of symptoms by 2 to 4 years of age, progression of somatic symptoms, severe cognitive impairment during childhood, and death by the teenage years. The attenuated type is associated with a later onset during childhood, a slower and milder progression of somatic disease, little to no cognitive impairment, and survival into adulthood3). Deletions and gross rearrangements of the IDS gene are associated with severe MPS II, whereas missense mutations are more often associated with the attenuated form4,-6).
IDS activity, which is measured in leukocyte pellets or fibroblasts, has been reported by Sukegawa-Hayasaka et al.7) to have a relationship with clinical phenotypes. However, no study about the relationship between plasma IDS activity and clinical phenotypes of MPS II patients has been conducted. If the relationship exists, we can hypothesize that the mild phenotype may result from some residual activities of the lysosomal enzymes. Our study is the first to describe the relationship between plasma IDS activity measured with a fluorimetric enzyme assay and MPS II clinical phenotypes.
Materials and methods
1. Subjects
We included 43 male patients with MPS II who were diagnosed at the Samsung Medical Center between April 1995 and May 2011. All patients or their parents and legal representatives provided written informed consent. The study was approved by the Institutional Review Board committee.
MPS II was tentatively diagnosed based on clinical findings as well as abnormal excretion of urinary glycosaminoglycan. Genetic tests as well as enzyme assays using leukocyte pellets were used to confirm the diagnosis of 43 patients. Mutations of IDS were identified in all cases. Clinical phenotypes of the patients were classified as severe if the patients had cognitive impairment, and as attenuated if no cognitive impairment was observed even with pronounced bone and visceral involvement. This classification was based on a report by Froissart et al.8) The cognitive function of patients was evaluated as 6 parts which include consciousness, orientation, speech (receptive, expressive), calculation, memory and judgment. If the patients had similar level of cognitive function of the age or orientation, language, calculation and memory to do normal living, they were classified as attenuated type. If not, as severe type.
2. Methods
Plasma IDS activity was measured with a fluorimetric enzyme assay using 4-methylumbelliferyl-╬▒-iduronate 2-sulphate according to the method by Voznyi et al.9). The reference range of plasma IDS activity was 167 to 475 nmol/4 hr/mL. For the standard IDS assay, reaction mixtures contained 10 ┬ĄL 5x diluted plasma to which 20 ┬ĄL 1.25 mmol/L MU-╬▒IdoA-2S in 0.1 mol/L sodium acetate buffer (pH 5.0) containing 10 mmol/L lead acetate and 0.02% sodium azide was added. The reaction mixtures were incubated for 4 hours at 37Ōäā.
Next, 20 ┬ĄL of concentrated McIlvain's buffer (pH 4.5; 0.4 mol/L Na-phosphate and 0.2 mol/L citrate) and 10 ┬ĄL (16 ┬Ąg protein) of partially purified ╬▒-iduronidase from rabbit liver (preparation essentially according to the method of Verheijen et al.10)) were added and the solution was incubated for 24 hours at 37Ōäā. Reactions were terminated by adding 200 ┬ĄL of 0.5 mol/L Na2CO3/NaHCO3 (pH 10.7). The fluorescence of 4-methylumbelliferone (MU) was measured with an Aminco-Bowman Series 2 (Thermo Electron Co., Madison, WI, USA).
3. Statistical analysis
Data were analyzed using SPSS ver. 18.0 (IBM Co., New York, NY, USA). The data were first analyzed using descriptive statistics (median and range). Due to the non-normal distribution, a non-parametric test (Mann-Whitney test) was used to compare age and plasma IDS activity between severe and attenuated type patients. Receiver-operator characteristics (ROC) curve analysis was performed to evaluate the predictive accuracy of plasma IDS activity for distinguishing severe and attenuated types. P values less than 0.05 were considered to be statistically significant.
Results
1. Patients
This study conducted on total 43 male patients with MPS II. Twenty-six patients (60.5%) were classified as having the severe type, and 11 patients (39.5%) were diagnosed as having the attenuated type. The median age of patients with the sever type was 9.5 years (range, 4 to 19 years), and 10 years (range, 4 to 34 years) for attenuated type patients. Patient age was not statistically different between patients with severe type and ones with the attenuated type (P=0.405). Clinical characteristics of the patients are presented in Table 1.
2. Plasma IDS activity
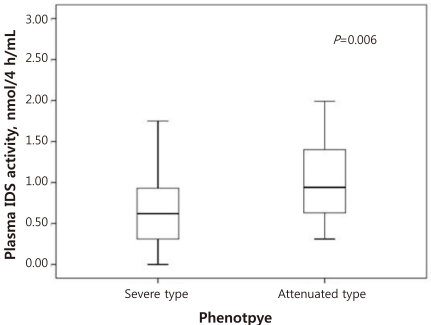
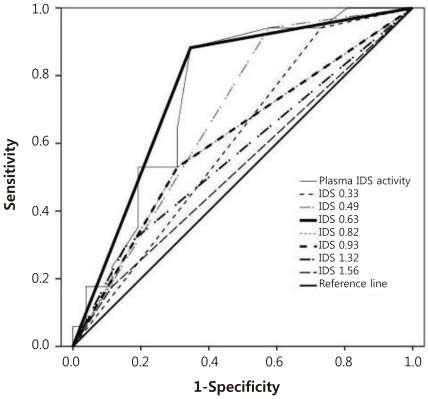
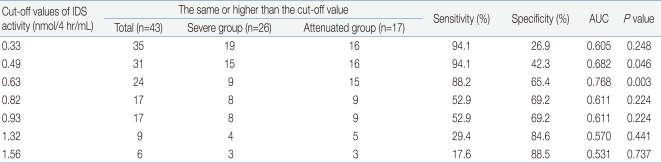
As shown Table 1, the median of plasma IDS activity was 0.62 nmol/4 hr/mL (range, 0 to 7.46 nmol/4 hr/mL) in for severe type patients and 0.94 nmol/4 hr/mL (range, 0.31 to 8.18 nmol/4 hr/mL) for patients with the attenuated type. Comparison of severe type and attenuated type patients showed statistically differences in plasma IDS activity. Plasma IDS activity (Table 1, Fig. 1) in individuals with the severe type was significantly lower than that of attenuated type patients (P=0.006). The optimal cut-off value of plasma IDS activity for distinguishing severe type from attenuated type was 0.63 nmol/4 hr/mL. This value had an 88.2% sensitivity, 65.4% specificity, and the area under ROC curve (AUC) was 0.768 (P=0.003) (Table 2, Fig. 2).
Discussion
MPS II is characterized by a wide phenotypic spectrum; cases of this disease can be classified into severe and attenuated types3). However, evaluation of clinical phenotypes is particularly difficult since patients are typically young at the time of diagnosis and there is no standardized index to score severity11). The relationship between IDS activity and clinical phenotypes was previously noted in 2006 by Sukegawa-Hayasaka et al.7). They measured IDS activity with the procedure of Hall et al.12) using radiolabelled disaccharide as the substrate. In 2001, Voznyi et al.9) developed a fluorimetric enzyme assay to measure IDS activity in fibroblasts, leukocytes, and plasma. Aside from being specific and sensitive, the IDS assay used in this study is less cumbersome and labor-intensive compared to assays using radiolabelled disaccharide. In particular, measuring residual plasma enzyme activity is a simpler method than examining skin fibroblasts or peripheral blood mononuclear cells. Therefore, the plasma IDS assay can be a useful method for evaluating MPS II patients. This is the first study showing the relationship between clinical phenotypes of MPS II patients and plasma IDS activity measured with a fluorimetric enzyme assay.
In this study, the plasma IDS activities of 43 male patients with MPS II were measured. Our study found a statistically significant difference in plasma IDS activities according to different clinical phenotypes of MPS II patients. Plasma IDS activity in severe type was significantly lower (P=0.006) than those with the attenuated type (Table 1, Fig. 1). In addition, the cut-off value of plasma IDS activity that could differentiate the severe type from the attenuated type was found to be 0.63 nmol/4 hr/mL with 88.2% sensitivity, 65.4% specificity, and an AUC of 0.768 (P=0.003) (Table 2, Fig. 2). In individuals with the severe type, 34.6% (9/26) of the patients had the same or higher plasma IDS activity than the cut-off value of 0.63 nmol/4 hr/mL; this was also observed in 88.2% (15/17) of patients with the attenuated type. Our findings suggest that patients with a plasma IDS activity less than 0.63 nmol/4 hr/mL are likely to be diagnosed with the severe type.
One limitation of this study was that the number of patients was relatively small. Our study also lacked a standardized scoring index of severity11). Furthermore, clinical phenotypes can change because the intellectual level of the patients can become more evident over time13). Therefore, a multicenter, large-scale study and the development of a standardized scoring system for classifying clinical phenotypes are needed.
In conclusion, the relationship between clinical phenotypes of MPS II patients and plasma IDS activity exits. Therefore we can hypothesize that the mild phenotype may result from some residual activities of the lysosomal enzymes.

 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


