

Introduction
Chronic granulomatous disease (CGD) is a group of rare X-linked or autosomal recessive genetic disorders of the phagocytic NADPH oxidase system causing recurrent bacterial and fungal infections with excessive inflammation and granuloma formation1, 2). Granuloma formation is considered to occur secondary to the failure of phagocytes in killing bacteria and due to undigested intracellular bacterial debris3).
CGD is normally diagnosed in infancy and has an incidence of 1 in 200,000-250,000 live births4, 5). Most patients manifest symptoms and signs of recurrent and persistent infections during the first 2 years of life. The disorder is much more common in males than in females, with 86% of patients in the USA national registry being male5). It has a mortality of approximately 2% per year, with a predicted life expectancy of around 25-30 years.
In this article, we describe an unusual case of aspergillus fumigatus pneumonia in a 2-month-old girl with CGD who showed a favorable response to treatment with amphotericin B and recombinant IFN-γ (r IFN-γ).
Case report
A 2 months old girl, born at a gestational age of 38+2 weeks was referred to us for investigations regarding persistent pneumonia. The girl suffered from several episodes of recurrent infections such as neonatal sepsis and gastroenteritis, since the 6th day of her life at the time of admission, she had received three recent course of antibiotics from the previous hospital including cefepime, clindamycin and imipenem due to persistent pneumonia. On admission, physical examination revealed mild chest retraction and rales in both lung fields. She was alert and responsive, with no neurological deficit. Her height and weight were at the 25th-50th percentile and the 50th-75th percentile for her age respectively.
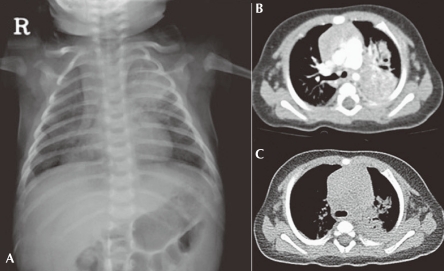
A chest X-ray (Fig. 1) confirmed the presence of bilateral pulmonary consolidation and atelectasis. Diagnostic bronchoalveolar lavage (BAL) was performed to obtain specimens for cytology and culture. A computed tomography (CT) of the thorax showed a left sided consolidation and evidence of hematogenously disseminated multifocal pneumonia.
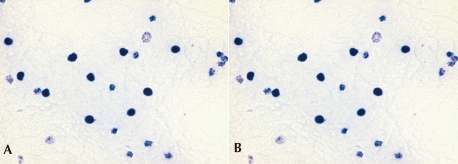
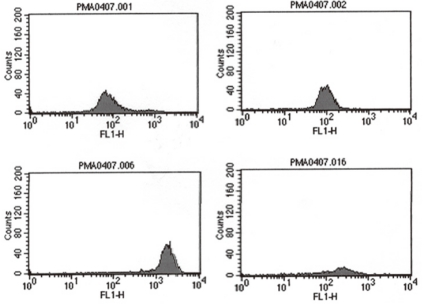
She had no family history related to primary immunodeficiency. Her total leucocyte count was 10,500/µL, with 34% neutrophils and 52% lymphocytes. The ESR was 12 mm/hr, and CRP 3.54 mg/dL. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels were raised (174 IU/L and 398 IU/L, respectively). Electrolyte levels and the results of renal function tests were normal. Investigations for her immunologic work-up showed Ig G, Ig A, Ig M, Ig E, C3, C4, and CH50 levels to be within her age specific reference range. Lymphocyte subset analysis revealed normal B cells and T cells for her age. Diagnosis of CGD was confirmed by both abnormal neutrophil nitroblue tetrazolium (NBT) slide test (Fig. 2) and neutrophil respiratory burst activity test (Fig. 3 B-1, B-2). The detection of Aspergillus galactomannan antigenemia during onceweekly monitoring was performed to facilitate early diagnosis of invasive aspergillosis and to evaluate the response to the antifungal therapy (amphotericin B). The course of the Aspergillus antigen galactomannan titer in the serum during treatment is shown in Fig. 4. A genetic study for the mutation of the NCF1 (Neutrophil cytosol factor 1) gene encoding the p47phox the most common gene mutation in autosomal recessive CGD, was done, but the mutation was not found.
After a positive result from BAL culture and Aspergillus antigen detection, an antifungal agent was added to the treatment. Amphotericin B therapy was initiated at a dose of 0.25 mg/kg/d, followed by a daily dosage increases of 0.25 mg/kg/d until a therapeutic dosage (1.0 mg/kg/d) was reached. However, the NBT dye reduction test revealed absolutely no reduction of dye by neutrophils therefore INF-γ administered subcutaneously at a dose of 15 µg three times weekly was added to the patient's regimen.
During the treatment for Aspergillus pneumonia, we found right-sided inguinal lymphadenopathy, at the previous central line insertion site, a subsequent culture yielded Enterobacter aerogenes. Perianal fissures had also occurred, and we therefore had the patient soak in a shallow tub of warm water (sitz bath). During the admission period, her AST and ALT levels rose significantly. We performed hepatitis serology and abdominal ultrasonography several times to identify the cause of the elevated liver enzymes. All tests produced normal results. We considered antibiotics-induced toxicity or the natural course of the disease as possible causes. We had added voriconazole to her regimen because of the residual lesions on the lung. However, during voriconazole administration, abnormal liver function values appeared, therefore we stopped voriconazole therapy.
At the time of discharge from our center, which was 3 months after admission, her acute phase inflammatory markers and Aspergillus precipitins had all progressively returned to normal. A chest radiograph and CT scan after a 3-month course of antifungal treatment with IFN-γ showed complete resolution of the previous findings. BAL culture and a serological test were both negative. However, six weeks after the discharge, she was readmitted with fever due to recurrent pneumonia, considered as a previous remnant lesion. She was treated with voriconazole for 4 weeks and commenced on prophylactic antibiotics and antifungal agents. We are currently considering HLA-identical bone marrow transplantation (BMT) for this patient at the age of 1-year-old.
Discussion
CGD is a primary immunodeficiency disorder characterized by defects in superoxide-generating systems of phagocytes leading to recurrent bacterial and fungal infections. Diagnosis of this disease requires a high index of suspicion along with prompt and aggressive treatment of infections. The presentation of this patient with recurrent, uncommon infections in early infanthood pointed towards a primary immunodeficiency. The patterns of presentation differ from patient to patient in CGD. As shown in this case, the lung is the most common site of infection, manifested as pneumonia or pulmonary abscess. Alternatively, dermatitis, subcutaneous abscess, lymphadenitis, liver abscess or osteomyelitis may be the prominent clinical problem in CGD patients7).
The diagnosis of CGD is confirmed by the NBT dye reduction test and the respiratory burst activity test. Normally the percentage of blue-stained neutrophils is close to 100, but in CGD patients, this percentage is close to 0. In this case, the neutrophil respiratory burst activity test showed a dual population in the mother of the patient, further her father's neutrophil respiratory burst activities were slightly decreased, which suggested an autosomal recessive inheritance pattern.
In CGD patients, neutrophils are able to phagocytose particles normally, but are unable to generate superoxide anions that are essential for microbial killing. The degree of superoxide anion production is determined partially according to the type of the inheritance pattern2). Therefore the functional and molecular diagnosis of CGD may be useful for predicting the prognosis and deciding the appropriate treatment. For example, there are several subtypes in gp91phox mutations, which are inherited in the pattern of X-linked form. In the X910, which accounting for approximately 90% of the X-linked CGD cases, NADPH oxidase activity is totally abolished because of both the instability of the mRNA and protein. X91-, accounting for less than 10% cases among the X-linked mutations, is a type of promoter mutation that causes 3-9% of normal NADPH oxidase activity, resulting in later diagnosis and a milder clinical course than X910. Autosomal recessive CGD, accounting for about 30-40% of all CGD cases is caused by the genetic defects in one of the p47phox, p67phox and p22phox.
The microorganisms responsible for the majority of infections in CGD are S. aureus, Gram-negative enteric bacilli (including Serratia marcescens, Salmonella species, and Burkholderia cepacia), and Aspergillus species. Aspergillus fumigatus infection as shown in this case, accounts for 15% of infections in CGD patients. It appears to occur because of the failure to clear hyphae by neutrophils' oxidant mechanisms. Further the defect in phagocytes' oxidant production is associated with a prolonged inflammatory response, causing granuloma formation3). The diagnosis of aspergillosis in CGD is difficult and important. In this case, we used noninvasive tests such as BAL culture and Aspergillus antigen detection without an invasive procedure such as lung tissue biopsy. Bronchoscopy with BAL is an established method for the diagnosis of pulmonary infections, especially in immunocompromised patients8). In a study, the diagnostic value sensitivity of bronchoscopy is 43% in the cases of histologically proven invasive pulmonary aspergillosis9). The Aspergillus galactomannan antigenemia assay may assist in the diagnosis of aspergillosis with a sensitivity of 81% and a specificity of 89%10).
Further, when amphotericin B is being used in therapy, falsenegative results may occur because the antifungal agent suppresses the growth of hyphae9). We monitored the antigenemia during once-weekly and observed declining values, suggesting successful treatment.
The management of CGD patients includes avoidance of infection with prophylactic antibiotic therapy and aggressive treatment of acute infections and inflammatory complications. In a randomized, non-blinded trial comparing voriconazole versus amphotericin B in the treatment of invasive aspergillosis, successful outcomes were noted in 52.8% of the patients in the voriconazole group and 31.6% of those in the amphotericin B group11). In our case, therapy with voriconazole during the first admission period had to be discontinued because of liver toxicity. When voriconazole was administered during the second admission period, it proved very effective and was well tolerated.
IFN-γ prophylaxis reduces the frequency of the serious infections by 70% by increasing the production of superoxides in CGD patients. Stem cell transplantation is curative in CGD9). Even in patients with active Aspergillus infection, human leukocyte antigen (HLA)-identical BMT or peripheral blood stem cell transplantation has been successfully performed with granulocyte-colony stimulating factor (G-CSF)-mobilized granulocyte transfusions10, 12). However in the absence of an HLA-identical family donor, BMT or peripheral blood stem cell transplantation is not recommended9). A therapeutic alternative for CGD patients with no HLA-identical donors is the genetic modification of autologous hematopoietic stem cells (HSC)13). Although CGD has been successfully corrected in animal models by gene transfer in HSCs, similar success have been achieved in humans with CGD. In this report, substantial gene transfer in CGD patients' neutrophils lead to a large number of functionally corrected phagocytes and notable clinical improvement13).

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


