 About
About Browse articles
Browse articles For contributors
For contributorsAll issues > Volume 69(5); 2026
Clinical application of whole exome and genome sequencing in pediatric neurodevelopmental disorders
-
Keun Soo Lee, MD1
 , Seung Hwan Oh, MD, PhD2, Ja Young Lee, MD, PhD3, Go Hun Seo, MD, PhD4, Da Eun Roh, MD5, Ji Kyoung Park, MD, PhD5, Bo Lyun Lee, MD, PhD5
, Seung Hwan Oh, MD, PhD2, Ja Young Lee, MD, PhD3, Go Hun Seo, MD, PhD4, Da Eun Roh, MD5, Ji Kyoung Park, MD, PhD5, Bo Lyun Lee, MD, PhD5
- Corresponding author: Bo Lyun Lee, MD, PhD. Department of Pediatrics, Busan Paik Hospital, Inje University College of Medicine, Bokji-ro 75, Busanjin-gu, Busan, 47392, Korea Email: bototii@paik.ac.kr
- Received November 21, 2025 Revised January 15, 2026 Accepted January 16, 2026
- Abstract
-
- Background
- Background
- Neurodevelopmental disorders (NDDs) are frequently encountered in pediatric neurology clinics. However, their extensive genetic heterogeneity often limits the diagnostic yield of standard diagnostic tests, highlighting the need for comprehensive genomic approaches.
- Purpose
- Purpose
- This study aimed to evaluate the diagnostic utility of whole exome sequencing (WES) and whole genome sequencing (WGS) in children with unexplained NDDs and assess the clinical relevance of the genomic findings.
- Methods
- Methods
- We retrospectively reviewed the medical records of 64 pediatric patients with NDDs who underwent WES or WGS between March 2018 and November 2024. Clinical data, neuroimaging and electroencephalography findings, and the results of previous genetic tests were analyzed. The diagnostic yield was calculated, and clinical characteristics were compared between patients with and without a genetically confirmed diagnosis. Patients were categorized as genetically confirmed (positive) when a definitive molecular diagnosis was identified through WES or WGS and as not genetically confirmed (negative) when no causative variant was detected.
- Results
- Results
- A definitive molecular diagnosis was achieved in 25 of 64 patients (39.1%). Diagnostic yields were 37.5% and 33.3% for WES and WGS, respectively. Most variants showed autosomal dominant (n=13) inheritance, followed by X-linked (n=9) and autosomal recessive (n=3) patterns. Novel variants accounted for 57.7% of the pathogenic or likely pathogenic variants. A positive family history was significantly associated with a higher diagnostic yield (20.0% vs. 2.6%, P=0.030), while prematurity was more common in the negative group (33.3% vs. 8.0%, P=0.032). Three WES-negative patients were later diagnosed using chromosomal microarray analysis (CMA) or repeat expansion testing.
- Conclusion
- Conclusion
- WES and WGS are effective diagnostic tools for pediatric NDDs. Phenotype re-evaluation and the selective use of genetic tests such as CMA and repeat expansion analysis enhance diagnostic yield.
- Introduction
- Introduction
Neurodevelopmental disorders (NDDs) represent a significant proportion of pediatric patients presenting to neurology clinics. These disorders encompass a broad spectrum of conditions, including intellectual disability (ID), global developmental delay (GDD), autism spectrum disorder (ASD), attention-deficit/hyperactivity disorder, and communication disorders, as well as conditions with neurological involvement associated conditions such as cerebral palsy and epilepsy [1]. Characterized by impairments in cognition, motor function, language, and behavior, NDDs exhibit considerable clinical and genetic heterogeneity, which presents substantial diagnostic challenges for clinicians [1,2].The initial diagnostic workup typically includes neuroimaging, standardized developmental assessments, electroencephalography (EEG), and specialized biochemical investigations, with test selection guided by the patient's specific clinical phenotype [3]. When a genetic etiology is suspected, chromosomal microarray analysis (CMA) and targeted single-gene or multigene panel testing are frequently employed as first-tier genetic investigations [4-6]. However, the extensive genetic heterogeneity underlying NDDs often limits the diagnostic yield of these approaches; CMA achieves diagnostic yields of approximately 10–20% and targeted gene panels demonstrate variable yields of 15%–30%, depending on patient selection and panel composition [4,5,7].Recent advances in molecular biology and bioinformatics have facilitated the clinical implementation of high-throughput sequencing technologies, particularly whole exome sequencing (WES) and whole genome sequencing (WGS) [8,9]. These genome-wide approaches have demonstrated superior diagnostic efficacy compared with traditional methods and are increasingly recognized as essential tools in the etiological investigation of unexplained NDDs. They significantly improve diagnostic yield, enhance prognosis prediction, and enable personalized management and comprehensive genetic counseling [8,10].The present study aimed to elucidate the diagnostic and clinical utility of WES/WGS in pediatric patients with NDDs evaluated at a specialized pediatric neurology clinic. This retrospective chart review focused on characterizing molecular diagnoses and associated clinical phenotypes to assess real-world diagnostic yield and clinical relevance of genomic testing in routine neurological practice.
- Methods
- Methods
- 1. Patients
- 1. Patients
A retrospective chart review was conducted on pediatric patients with NDDs who had undergone WES and/or WGS at the Pediatric Neurology Clinic of Busan Paik Hospital between March 2018 and November 2024. Patients were included if they met clinical diagnostic criteria for one or more NDDs including GDD, ASD, ID, and epilepsy accompanied by developmental delay or ID, and remained without a definitive etiology after standard clinical and diagnostic evaluations.GDD was defined as a delay in 2 or more developmental domains (gross motor, fine motor, social and personal, language, and cognition) in young children, as assessed using validated developmental tools, including the Korean Developmental Screening Test for Infants and Children and the Bayley Scales of Infant and Toddler Development, Third Edition [11,12]. ID was defined as an IQ score of less than 70 in children aged 5 years or older, with concurrent impairments in cognitive functioning and adaptive behavior, as measured by the Korean Wechsler Intelligence Scale [12,13]. ASD was diagnosed according to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition criteria [13]. Only patients with epilepsy in whom seizures were accompanied by developmental delay or ID were included; patients with isolated epilepsy without neurodevelopmental impairment were excluded.All patients underwent comprehensive clinical evaluations, including neurologic examination, standardized developmental assessments, and neuroimaging. WES or WGS was performed in patients with unexplained NDDs after inconclusive results from prior diagnostic investigations. The decision to proceed with genomic sequencing was made by pediatric neurologists based on phenotype severity, clinical complexity, and the absence of an established molecular diagnosis.All participants and their families received formal pretest genetic counseling, which included a detailed explanation of the potential benefits, limitations, and risks associated with genomic testing. Informed consent for genetic analysis was obtained in accordance with institutional guidelines. This study was approved by the Institutional Review Board of Busan Paik Hospital (approval No. BPIRB 2024-11-011).- 2. Clinical data collection
- 2. Clinical data collection
Clinical data, including demographic characteristics, phenotypic features, laboratory results, EEG findings, and brain magnetic resonance imaging (MRI) results, were systematically collected from electronic medical records. Family history was defined as the presence of a definite history of NDDs in first- or second-degree relatives. Phenotypes of interest included GDD, epilepsy, facial dysmorphism, and other neurological or syndromic features. Abnormal growth and head circumference were defined as measurements below or above 2 standard deviations from the mean for age- and sex-matched populations. Multiple anomalies are defined as the presence of 2 or more major congenital malformations affecting different organ systems.- 3. Genetic testing strategy
- 3. Genetic testing strategy
Prior diagnostic evaluation included genetic and metabolic investigations, such as conventional karyotyping, CMA, FMR1 gene testing for fragile X syndrome, methylation-specific polymerase chain reaction (PCR) for Prader-Willi and Angelman syndromes, MECP2 gene analysis, targeted multigene panels, and relevant biochemical assays.WES and WGS were implemented as second- or third-tier diagnostic tests following inconclusive results from earlier investigations. Some patients underwent WES as a first-line test based on clinical urgency, phenotype severity, or test accessibility. In cases where WES did not yield a definitive molecular diagnosis, additional genetic tests were performed. CMA was selectively performed when clinically indicated, especially in cases where copy number variant (CNV) analysis was not sufficiently captured by initial sequencing and CMA had not been conducted before WES. Additional targeted assays, such as trinucleotide repeat expansion testing, were performed when indicated by specific clinical features identified after rephenotyping.- 4. WES and WGS
- 4. WES and WGS
Genomic DNA was extracted from peripheral blood leukocytes for WGS and from buccal swabs for WES for each proband. WES was performed at 3billion, Inc. (Korea) using the IDT xGen Exome Research Panel v2 (Integrated DNA Technologies, USA) for exome capture. Sequencing was performed using the Illumina NovaSeq 6000 platform (Illumina, USA). WGS was performed by Macrogen, Inc. (Korea). DNA libraries were prepared using the TruSeq DNA PCR-free sample preparation kit (Illumina), and sequencing was performed using the Illumina NovaSeq 6000 system. WGS sequencing data analysis was performed at 3billion, Inc. to identify a comprehensive range of variants, including single-nucleotide variants, small insertions/deletions, CNVs, structural variants, repeat expansions, mobile element insertions, and regions of homozygosity [14,15].Variants were annotated, filtered, and prioritized using EVIDENCE, 3billion’s proprietary system with a daily-updated database, variant classification module, and symptom similarity scoring module [14]. The variants were classified according to the guidelines of the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) into 5 categories: pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign, or benign [16-18]. The candidate variants were manually reviewed by clinical geneticists and selected for reporting based on their correlation with the patient’s phenotype.It should be noted that earlier WES analyses performed during 2018–2020 were based on capture-based exome sequencing platforms and analytical pipelines that were primarily optimized for single-nucleotide variants and small insertions/deletions. As such, the sensitivity for detecting CNVs, repeat expansions, and certain structural variants was limited, and these variant classes were not systematically evaluated at that time.Sanger sequencing was performed for variant confirmation when sufficient DNA sample was available. Parental and familial segregation analyses were performed whenever possible. For the purposes of analysis, patients were classified as “positive” if a causative variant was identified by WES or WGS, and as “negative” if no causative variant was identified by these sequencing methods (Table 1). Novel variants were defined as those absent from ClinVar and without prior disease-associated reports in the literature at the time of analysis.- 5. Statistical analyses
- 5. Statistical analyses
All statistical analyses were performed using IBM SPSS Statistics ver. 29.0 (IBM Co., USA). Continuous variables were reported as means with standard deviations, whereas categorical variables were expressed as percentages. For comparison of continuous variables, Student t test was employed. Categorical variables were analyzed using Pearson chi-square test or Fisher exact test, with the latter applied when the expected cell frequency was <5. Two-tailed null hypotheses of no difference were rejected if the P values were <0.05.
- Results
- Results
- 1. Patients’ clinical characteristics
- 1. Patients’ clinical characteristics
A total of 70 patients were enrolled during the study period. After excluding individuals with infantile seizures, paroxysmal dyskinesia, or skeletal deformities in the absence of neurodevelopmental delay, 64 patients (30 males [46.9%], 34 females) were included in the final analysis (Fig. 1). The mean age at symptom onset (mean±standard deviation) was 1.2±2.0 years, and the mean age at WES or WGS testing was 4.6±5.0 years (Table 1).The most prevalent clinical phenotype was GDD, observed in 57 of the 64 patients (89.1%). Epilepsy was present in 25 patients (39.1%) and ASD in one (1.6%) (Table 1). Hypotonia was noted in 32 patients (50.0%) and short stature in 24 (37.5%). Abnormal movements, including ataxia, tremor, and myoclonus, were reported in 5 patients (7.8%).Brain MRI was performed in 62 patients, of whom 33 (53.2%) demonstrated structural abnormalities, including cerebellar dysplasia, reduced cerebral white matter volume, cerebellar vermis hypoplasia, dysgenesis or agenesis of the corpus callosum, diffuse cerebral atrophy, ventriculomegaly, Dandy-Walker variant, white matter injury, cortical dysplasia, and polymicrogyria. EEG was conducted in 43 patients, of whom 22 (51.2%) demonstrated abnormal findings, including focal epileptiform discharges, background slowing, and generalized slow-wave activity. An overview of the genetic investigations conducted in our cohort is presented in Table 1.- 2. Diagnostic yield of WES/WGS
- 2. Diagnostic yield of WES/WGS
WES was performed in 56 patients and WGS in 12 (Fig. 1). Among those who underwent WES, a molecular diagnosis was achieved in 21 individuals (37.5%). Of the 12 patients who underwent WGS, 4 (33.3%) were diagnosed with a pathogenic or likely pathogenic variant. WES was performed as a first-tier test in 6 patients (patient numbers 4, 6, 11, 14, 17, and 24) of these positive cases. Of the 39 patients who remained negative, 10 had undergone WES as an initial investigation. Among the 4 individuals with negative WES results, subsequent WGS revealed a pathogenic variant in 1 patient, leading to a genetic diagnosis. Twelve patients, along with 2 affected siblings and 23 unaffected family members, underwent WGS as part of the National Project of Bio Big Data. Among these, 10 families were enrolled as proband-parent trios, one family as a proband-sibling-parent quartet, and one family as a proband-sibling-single-parent trio. All patients who underwent WGS had previously received genetic evaluations, including WES or other genetic tests such as conventional karyotyping, CMA, FMR1 gene testing for fragile X syndrome, MECP2 gene analysis, and targeted multigene panels. Periodic reanalysis of WES/WGS data was not performed in this study.Overall, 25 of the 64 patients (39.1%) received a molecular diagnosis through WES or WGS. Of the variants identified through WES/WGS, 16 were classified as pathogenic, 10 as likely pathogenic, and one as a VUS (Table 2). Of the 25 genetically and clinically confirmed diagnoses, inheritance patterns included autosomal dominant in 13 cases, X-linked in 9 cases, and autosomal recessive in 3 cases. Sanger sequencing in 21 probands and their family members confirmed 14 de novo variants. Notably, 15 of the pathogenic or likely pathogenic variants were novel, 7 of which had been previously reported in our earlier study [19-21]. Comprehensive phenotypic data of patients with pathogenic or likely pathogenic variants are provided in Supplementary Table 1. Detailed information on population allele frequencies, ClinVar reporting status, and the ACMG/AMP criteria applied for variant classification is summarized in Supplementary Table 2.- 3. Representative cases with diagnostic implications
- 3. Representative cases with diagnostic implications
Patient 19 harbored a novel pathogenic variant presented with distinctive clinical features. The patient was born at 40 weeks and 5 days of gestation with a birth weight of 3.06 kg. At 9 months of age, the patient developed focal impaired awareness seizures characterized by altered consciousness, cyanosis, and vomiting, with episodes lasting >1 hour, leading to a diagnosis of epilepsy. The patient exhibited GDD and progressive microcephaly, with head circumference declining to below the 3rd percentile, despite being within the normal range at birth. Dysmorphic facial features included synophrys, hypertelorism, narrow nasal bridge, bulbous nasal tip, and thin upper lip. Brain MRI revealed ventricular dilatation and mild cerebral atrophy (Fig. 2A and B).Initial genetic evaluations, including conventional karyotyping, CMA, MECP2 gene sequencing, and an epilepsy gene panel, failed to identify a causative variant. Subsequent WGS identified a novel heterozygous missense pathogenic variant in CDC42 (NM_001791.4:c.67T>C; p.Tyr23His), leading to a diagnosis of Takenouchi-Kosaki syndrome. This is the first reported case of Takenouchi-Kosaki syndrome in Korea. The variant was confirmed to be de novo, and appropriate genetic counseling was provided to the family. The patient was also registered in the Korean National System for Newly Identified Ultra-Rare Disorders.A case of clinical interest is that of patient number 23, a female infant born at term. Prenatal ultrasonography had revealed corpus callosum hypoplasia and mega cisterna magna. Postnatal MRI confirmed corpus callosum hypoplasia and periventricular nodular heterotopia (Fig. 2C and D). Initial genetic investigations, including conventional karyotyping and CMA, performed during admission to the neonatal intensive care unit, were unremarkable. Because of the accompanying developmental delay, WES was performed, which identified a VUS in FLNA (NM_001456.4:c.7895G>T; p.Ser2640Ile). Although neither parent exhibited overt clinical features suggestive of an X-linked disorder, a segregation analysis was conducted under the presumption of a potential de novo variant. However, an identical variant was detected in the asymptomatic mother. Upon further clinical enquiry, the mother disclosed a history of mild developmental delay. A subsequent brain MRI revealed periventricular nodular heterotopia, consistent with the daughter’s neuroimaging findings. Based on phenotypic concordance and segregation evidence, the variant was reclassified as likely pathogenic, establishing a molecular diagnosis.- 4. Comparative analysis of positive versus negative patients
- 4. Comparative analysis of positive versus negative patients
A comparison between patients who were genetically diagnosed using WES/WGS and those who remained negative revealed notable clinical differences (Table 1). Among the 64 patients included in this study, 25 (39.1%) received a definitive molecular diagnosis via WES or WGS.The proportion of male patients was lower in the positive group than in the negative group (32.0% vs. 56.4%), although the difference was not significant (P=0.074). A positive family history of NDDs was more common in the positive group (20.0%) than in the negative group (2.6%, P=0.030), whereas a history of prematurity was more frequent in the negative group (33.3% vs. 8.0%, P=0.032). Among WES/WGS-positive cases, 3 patients had a family history of GDD/ID, and 2 had a family history of epilepsy accompanied by GDD/ID. Among WES/WGS-negative cases, 1 patient had a family history of epilepsy accompanied by GDD.There were no significant differences between groups regarding age at symptom onset, timing of WES/WGS, or major clinical features such as developmental delay, epilepsy, microcephaly, hypotonia, short stature, facial dysmorphism and multiple anomalies. Neuroimaging and EEG abnormalities were comparably distributed between the groups, and no significant difference was observed in the use of WES or WGS.- 5. Follow-up genetic testing in WES-negative cases
- 5. Follow-up genetic testing in WES-negative cases
Among the cohort of patients who remained negative following WES, 3 individuals were later diagnosed through CMA or targeted genetic testing.The first case involved an 8.7-year-old girl with epilepsy, GDD, dysmorphic facial features, hypotonia, and intellectual disabilities. CMA performed 2 months after nondiagnostic WES revealed a 4.5-Mb deletion at 1q44 (chr1:244,737,375–249,224,684), consistent with 1q44 microdeletion syndrome. The second case involved an 11.8-year-old girl with epilepsy, GDD, dysmorphic features, and short stature. WES failed to identify pathogenic variants, but subsequent CMA detected a 10-Mb duplication at 15q11.2–q13.3 (chr15:22,770,421–32,915,723) involving 4 copies, leading to a diagnosis of 15q duplication syndrome. Importantly, both patients had undergone prior genetic evaluations, including conventional karyotyping, Prader-Willi/Angelman syndrome methylation-specific PCR, or targeted gene panel testing, all of which failed to yield a diagnosis prior to WES. The third patient was a 14.5-year-old boy with GDD, ID, dysmorphic features, spasticity, cerebral palsy, and macrocephaly. Brain MRI revealed bilateral lateral ventricular dilatation and suspected white matter volume loss. Despite negative PTEN gene analysis and WES results, further clinical evaluation revealed a high-arched palate, stiff tongue, and suspected handgrip myotonia prompting DMPK trinucleotide repeat analysis, which confirmed >150 CTG repeats, establishing a diagnosis of childhood-onset myotonic dystrophy type 1. In this case, myotonic dystrophy was not initially suspected, as the patient did not exhibit classic clinical features of the disorder at the time of the initial evaluation. The patient had a history of prematurity and periventricular leukomalacia, which likely contributed to central nervous system manifestations, including spasticity, and may have confounded the initial phenotypic interpretation. These cases underscore the importance of continued phenotypic re-evaluation and the utility of sequential genetic testing beyond WES in pediatric patients with complex neurodevelopmental phenotypes.
- Discussion
- Discussion
Our findings demonstrate that genome-wide sequencing provides a meaningful diagnostic yield in pediatric NDDs, consistent with prior studies reporting yields of approximately 30%–50% [2,8,9,22]. Of note, a positive family history of NDDs was significantly associated with a higher diagnostic yield, whereas prematurity was more frequently observed among negative patients, suggesting the influence of non-genetic or perinatal factors.A notable proportion of the variants identified in our cohort were novel ones, which highlights that discovery and characterization of genetic variants in NDDs is an ongoing process. More than half of the pathogenic or likely pathogenic variants had not been previously reported, with 15 of 26 variants (57.7%) classified as novel. This observation is consistent with findings from other cohorts, in which 40–60% of disease-causing variants were classified as novel [9,23]. Such results reflect both the limitations of existing variant databases and the strength of exome/genome sequencing in rare disease discovery.Autosomal dominant inheritance emerged as the predominant pattern among the confirmed diagnoses, consistent with previous studies emphasizing the major contribution of de novo autosomal dominant variants to the etiology of NDDs [9,23]. These findings highlight the diagnostic value of trio-based approaches in detecting de novo variants, as observed in a substantial fraction of our positive cases.A meta-analysis by Srivastava et al. [22] demonstrated that WES consistently outperforms CMA as a first-tier diagnostic test for unexplained NDDs, with diagnostic yields of approximately 36% versus 15%–20% for CMA. Moreover, the identification of novel variants in over half of the diagnosed cases in our cohort highlights the value of WES and WGS in elucidating rare genetic etiologies [9,23]. These findings support a structured, stepwise genomic diagnostic approach for pediatric patients with NDDs in whom a specific or clinically suspected monogenic disorder has not been identified after initial clinical evaluation. In line with recent recommendations of the American Academy of Pediatrics [24], the ACMG guidelines [8], and previous meta-analytic evidence [22], we outline a literature-based diagnostic framework that prioritizes early implementation of genome-wide sequencing with integrated CNV analysis, followed by the selective use of complementary modalities rather than routine sequential testing (Fig. 3). In this context, CMA may be considered when CNV analysis is not adequately captured by the initial sequencing approach. For patients who remain negative on WES or WGS, additional steps described in the literature include phenotypic re-evaluation, additional targeted testing (e.g., fragile X testing, imprinting disorders, repeat expansion disorders, mitochondrial DNA testing, and metabolic evaluation for inborn errors of metabolism), and periodic reanalysis of sequencing data, which has been shown to improve the diagnostic yield as variant interpretation frameworks evolve [22,24]. Although such reanalysis has demonstrated clinical utility, systematic reanalysis was not performed in this study due to the retrospective design and limitations in data availability and analytical resources.Importantly, the final interpretation of genetic testing results should not rely solely on laboratory-based variant classification but should be integrated with expert clinical assessment including detailed phenotypic reassessment, family history, and segregation analysis, to accurately determine variant pathogenicity and clinical relevance [22,24]. Identification of a de novo CDC42 variant enabled diagnosis of Takenouchi-Kosaki syndrome, expanding its phenotypic spectrum. In an FLNA-related case, maternal phenotyping and MRI enabled reclassification of an uncertain variant to likely pathogenic. These findings emphasize that WES and WGS achieve maximal yield when combined with careful phenotypic assessment and family studies. In addition, selective application of sequential diagnostic modalities remained important, as CMA detected clinically relevant CNVs in exome-negative cases, consistent with previous evidence supporting CNV analysis as a complementary tool [8,22,24].Our results also highlight key clinical correlations of diagnostic success. Patients with a family history of NDDs were more likely to receive a genetic diagnosis, consistent with previous findings that emphasize the utility of trio-based analyses in familial cases. Conversely, premature birth was associated with a lower genetic diagnostic yield in some cohorts, suggesting a greater role for non-genetic or multifactorial etiologies. In a meta-analysis of 15 exome sequencing cohorts of cerebral palsy, the diagnostic yield was markedly higher in cryptogenic cases (35%) than in noncryptogenic cases defined by perinatal risk factors, including prematurity (7%) [2]. Similarly, in another exome sequencing cohort, a reduced likelihood of monogenic diagnoses was observed in the noncryptogenic subgroup characterized by prematurity (≤32 weeks) and other perinatal insults [25].This study has some limitations. First, this was a retrospective chart review conducted at a single tertiary care center, which may limit the generalizability of the findings. Second, trio-based WES was not routinely performed; therefore, confirming de novo variants and interpreting variants of uncertain significance were not always possible. Third, resource limitations and the high cost of genomic testing precluded the uniform application of WES or WGS across the cohort. Testing decisions were influenced by clinical urgency, test accessibility and financial feasibility, potentially introducing selection bias and limiting the assessment of diagnostic yield. Fourth, the relatively small number of patients who underwent WGS limited the evaluation of its incremental diagnostic yield. Finally, CMA was not systematically performed owing to partial insurance coverage and associated costs, which may have resulted in missed diagnoses related to CNVs in WES-negative cases.Despite these limitations, this study reinforces the role of genome-wide sequencing in pediatric neurogenetics and highlights the need for ongoing phenotypic assessments and multimodal testing strategies. Future prospective studies incorporating trio-based designs and standardized genomic testing protocols are warranted to validate and extend these findings.In conclusion, our findings demonstrate that while WES and WGS are highly effective diagnostic tools, their full clinical utility is achieved when integrated into a comprehensive phenotype-driven diagnostic framework. These findings support an iterative, comprehensive diagnostic approach in pediatric neurology, to enhance clinical decision-making, genetic counseling, and rare disease identification. Despite the limitations of this study, the findings reinforce the clinical utility of genome-wide sequencing and highlight the need for standardized, accessible genomic testing strategies to optimize care for children with unexplained NDDs.
Supplementary materials
Supplementary materials
Supplementary Tables 1-2 are available at https://doi.org/10.3345/cep.2025.02775.Supplementary Table 1.
cep-2025-02775-Supplementary-Table-1.pdfDetailed phenotypic characteristics of patients with pathogenic or likely pathogenic variants identified by whole exome/genome sequencingSupplementary Table 2.
cep-2025-02775-Supplementary-Table-2.pdfPopulation frequency, ClinVar status, and ACMG/AMP criteria for variants identified by whole exome/ genome sequencing
- Footnotes
-
Conflicts of interest No potential conflict of interest relevant to this article was reported.
Funding This work was supported by a 2023 research grant from Inje University.
Acknowledgments The authors thank Ms. Dahye Kim, the coordinator of the Busan Regional Center for Rare Diseases, for her assistance in patient recruitment and sample collection. We are also grateful to all patients and their families for their participation, which made this study possible. Whole genome sequencing was performed as part of the National Project for BioBig Data.
Author Contribution Conceptualization: JYL, BLL; Formal analysis: KSL, SHO, GHS, DER, JKP, BLL; Investigation: KSL, DER, JKP, BLL; Methodology: SHO, JYL, GHS, BLL; Project Administration: KSL, JYL, DER, JKP, BLL; Writing – Original Draft: KSL, SHO, JYL, DER, JKP, BLL; Writing – Review & Editing: KSL, SHO, JYL, GHS, DER, JKP, BLL
-
Fig. 1.
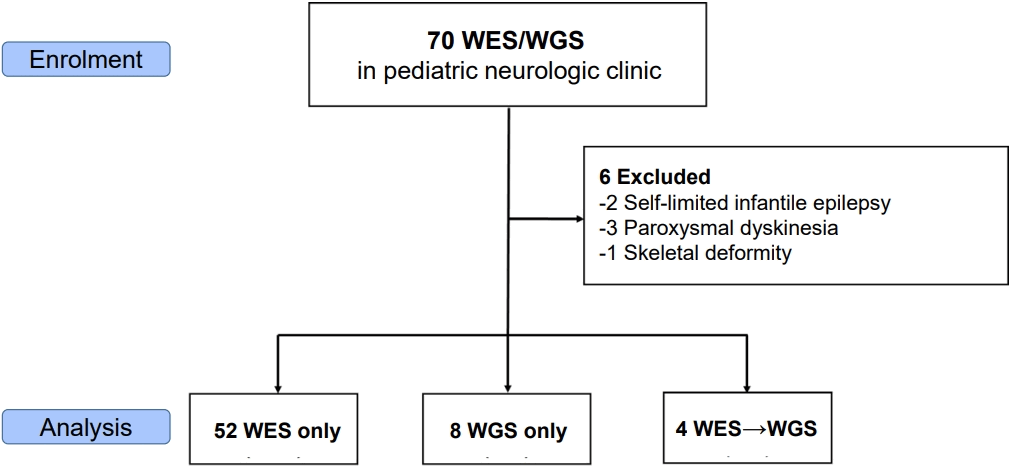
Fig. 2.
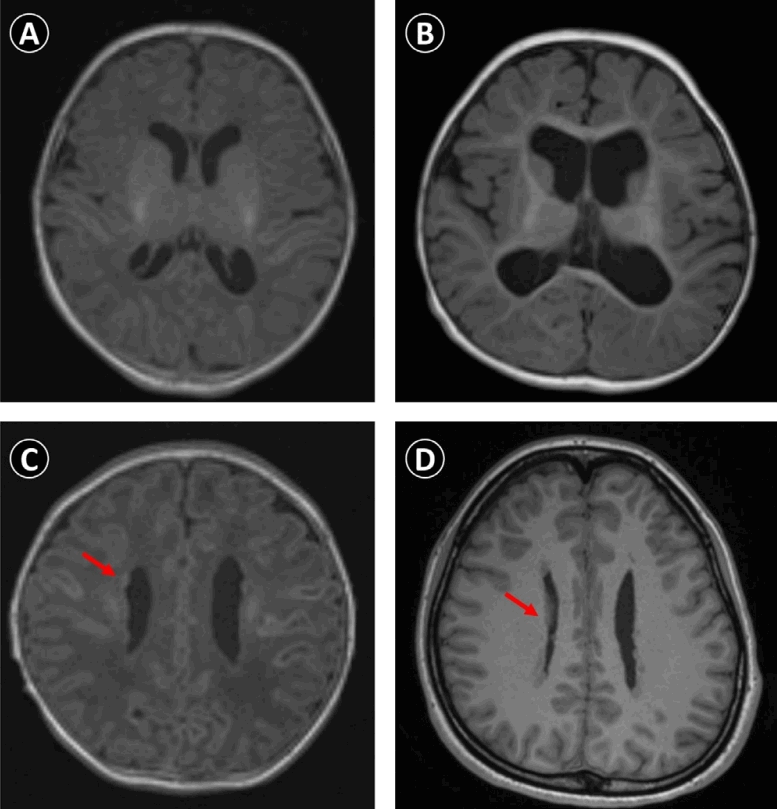
Fig. 3.
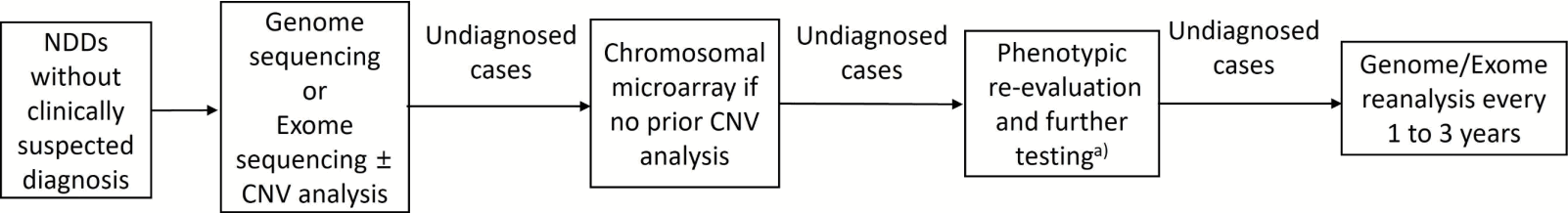
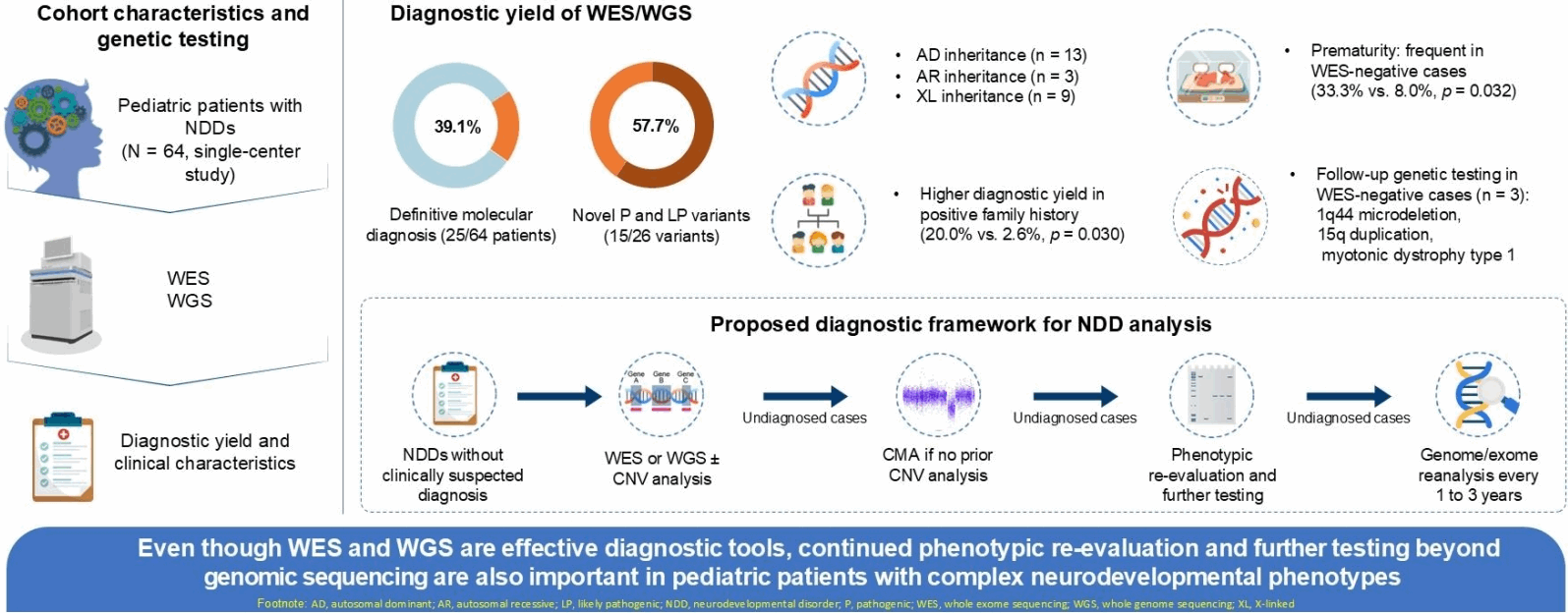
Table 1.
| Variable | Positive (n=25) | Negative (n=39) | Total (n=64) | P value |
|---|---|---|---|---|
| Age at testing (yr) | 4.4±5.3 | 4.8±4.9 | 4.6±5.0 | 0.691 |
| Male sex | 8 (32.0) | 22 (56.4) | 30 (46.9) | 0.074 |
| Age at symptom onset (yr) | 1.4±2.6 | 1.1±1.5 | 1.2±2.0 | 0.580 |
| Age at diagnosis (yr) | 4.4±5.3 | |||
| Time to diagnose (yr) | 3.0±3.9 | |||
| Past medical history | ||||
| Prematurity | 2 (8.0) | 13 (33.3) | 15 (23.4) | 0.032 |
| Perinatal problem | 6 (24.0) | 14 (35.9) | 20 (31.3) | 0.411 |
| Family history of NDD | 5 (20.0) | 1 (2.6) | 7 (9.4) | 0.030 |
| Clinical phenotype | ||||
| Developmental delay | 24 (96.0) | 33 (84.6) | 57 (89.1) | 0.231 |
| Epilepsy | 11 (44.0) | 14 (35.9) | 25 (39.1) | 0.603 |
| Autism spectrum disorder | 0 (0.0) | 1 (2.6) | 1 (1.6) | 1.000 |
| Microcephaly | 10 (40.0) | 13 (33.3) | 23 (35.9) | 0.605 |
| Macrocephaly | 2 (7.7) | 3 (8.0) | 5 (7.8) | 1.000 |
| Hypotonia | 14 (56.0) | 18 (46.2) | 32 (50.0) | 0.609 |
| Short stature | 11 (44.0) | 13 (33.3) | 24 (37.5) | 0.436 |
| Facial dysmorphism | 17 (68.0) | 21 (53.8) | 38 (59.4) | 0.305 |
| Abnormal movements | 2 (8.0) | 3 (7.7) | 5 (7.8) | 1.000 |
| ADHD | 1 (4.0) | 1 (2.6) | 2 (3.1) | 1.000 |
| Multiple anomalies | 11 (44.0) | 17 (43.6) | 28 (43.8) | 0.974 |
| Workup | ||||
| MRI-detected brain anomalies | 13/24 (54.2) | 20/38 (52.6) | 33/62 (53.2) | 1.000 |
| Abnormal EEG | 11/19 (57.9) | 11/24 (45.8) | 22/43 (51.2) | 0.543 |
| Genetic studies performed | ||||
| Karyotyping | 18 (72.0) | 27 (69.2) | 45 (70.3) | 1.000 |
| Chromosomal microarray | 9 (36.0) | 20 (51.3) | 29 (45.3) | 0.305 |
| FMR1 gene | 3 (12.0) | 3 (7.7) | 6 (9.4) | 0.671 |
| PWS/AS methylation PCR | 0 (0) | 4 (10.3) | 4 (6.2) | 0.149 |
| MECP2 gene | 1 (4.0) | 1 (2.6) | 2 (3.1) | 1.000 |
| Epilepsy gene panel | 1 (4.0) | 1 (2.6) | 2 (3.1) | 1.000 |
| Prior genetic testinga) | 19 (76.0) | 29 (74.4) | 48 (75.0) | 0.882 |
| WES | 22 (88.0) | 34 (87.2) | 56 (87.5) | 1.000 |
| WGS | 4 (16.0) | 8 (20.5) | 12 (18.8) | 0.751 |
Values are presented as mean±standard deviation or number (%).
NDD, neurodevelopmental disorder; WES, whole exome sequencing; WGS, whole genome sequencing; ADHD, attention-deficit/hyperactivity disorder; MRI, magnetic resonance imaging; EEG, electroencephalography; PWS/AS, Prader-Willi syndrome/Angelman syndrome; PCR, polymerase chain reaction.
Table 2.
| Pt. No. | Sex | Age (yr) | Gene | Reference transcript | Nucleotide change | HGVS protein change | Zygosity | Class | Inheritance | Variant origin | OMIM phenotype (MIM#) | Reference (PMID) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 2.3 | ARID1B | NM_020732.3 | c.2201dupG | p.(Ser736Ilefs*27) | Het | P | AD (novel) | De novo | Coffin-Siris syndrome (135900) | 32161024a), 37795942a) |
| 2 | M | 2.4 | SLC9A6 | NM_001330652.2 | c.675_676ins100b) | p.(Asp226Serfs*2) | Hem | LP | XL (novel) | Unknown | Intellectual developmental disorder, X-linked syndromic, Christianson type (300243) | 37795942a) |
| 3 | F | 9.3 | SYNGAP1 | NM_006772.3 | c.1715G>A | p.(Trp572*) | Het | LP | AD (novel) | Unknown | Intellectual developmental disorder, autosomal dominant 5 (612621) | 37795942a) |
| 4 | F | 9.4 | SCN2A | NM_021007.3 | c.4978T>G | p.(Leu1660Val) | Het | LP | AD (novel) | De novo | Developmental and epileptic encephalopathy 11 (613721) | 37795942a) |
| 5 | M | 8.2 | NSD1 | NM_022455.5 | c.5798A>G | p.(Asn1933Ser) | Het | LP | AD | Maternal | Sotos syndrome (117550) | 37795942a) |
| 6 | F | 0.5 | DCX | NM_000555.3 | c.358C>T | p.(Arg120*) | Het | P | XL | De novo | Lissencephaly, X-linked (300067) | 11175293, 37795942a) |
| 7 | M | 7.8 | SLC6A8 | NM_005629.4 | c.626_627delCT | p.(Pro209Argfs*87) | Hem | P | XL | Maternal | Cerebral creatine deficiency syndrome (300352) | 32434645, 37795942a) |
| 8 | M | 5.6 | CSNK2A1 | NM_177559.3 | c.593A>G | p.(Lys198Arg) | Het | LP | AD | Maternal | Okur-Chung neurodevelopmental syndrome (617062) | 37795942a), 25741868 |
| 9 | M | 1.3 | TAF1 | NM_001286074.2 | c.3701G>A | p.(Arg1234Gln) | Hem | LP | XL (novel) | Maternal | Intellectual developmental disorder, X-linked syndromic 33 (300966) | 37795942a) |
| 10 | F | 11.3 | NR2F1 | NM_005654.6 | c.983dupT | p.(Thr329Hisfs*68) | Het | LP | AD (novel) | Unknown | Bosch-Boonstra-Schaaf optic atrophy syndrome (615722) | 37795942a) |
| 11c) | F | 0.5 | NF1 | NM_001042492.3 | c.5812+332A>G | p.(?) | Het | P | AD | De novo | Neurofibromatosis, type 1 (162200) | 8829638, 18546366 |
| 12 | F | 0.1 | FLNA | NM_001110556.2 | c.2752dup | p.(Asp918Glyfs*13) | Het | P | XL | De novo | Heterotopia, periventricular, 1 (300049) | NA |
| 13 | F | 4.3 | DYRK1A | NM_001396.4 | c.665-3C>G | p.(?) | Het | LP | AD (novel) | De novo | Intellectual developmental disorder, autosomal dominant 7 (614104) | NA |
| 14 | F | 1.0 | CASK | NM_003688.3 | c.535del | p.(Arg179Valfs*22) | Het | P | XL (novel) | De novo | Intellectual developmental disorder and microcephaly with pontine and cerebellar hypoplasia (300749) | 35777792a) |
| 15c) | F | 0.1 | NEB | NM_001271208.2 | c.[5364G>A]; [21623G>T]d) | p.(Trp1788*); (Ser7208Ile) | Hete) | P/LP | AR (novel)e | Paternal/maternal | Nemaline myopathy 2, autosomal recessive (256030) | 25205138 |
| 16 | F | 2.6 | MED12 | NM_005120.3 | c.3443G>A | p.(Arg1148His) | Het | P | XL | De novo | Opitz-Kaveggia syndrome (305450) | 23395478, 24715367 |
| 17 | M | 2.0 | CACNA1A | NM_001127222.2 | c.3134C>G | p.(Ser1045*) | Het | P | AD (novel) | De novo | Developmental and epileptic encephalopathy 42 (617106) | NA |
| 18 | M | 0.1 | CREBBP | NM_004380.3 | c.1270C>T | p.(Arg424*) | Het | P | AD | De novo | Rubinstein-Taybi syndrome 1 (180849) | 16021471 |
| 19c) | F | 0.9 | CDC42 | NM_001791.4 | c.67T>C | p.(Tyr23His) | Het | P | AD (novel) | De novo | Takenouchi-Kosaki syndrome (616737) | NA |
| 20 | F | 0.3 | LAMA1 | NM_005559.4 | c.4252_4255dup | p.(Cys1419*) | Hom | P | AR (novel) | De novo | Poretti-Boltshauser syndrome (615960) | NA |
| 21c) | M | 18.8 | SLC6A8 | NM_005629.4 | c.1395_1397del | p.(Gly466del) | Hem | P | XL (novel) | Maternal | Cerebral creatine deficiency syndrome 1 (300352) | NA |
| 22 | F | 0.2 | PTPN11 | NM_002834.5 | c.922A>G | p.(Asn308Asp) | Het | P | AD | De novo | Noonan syndrome 1 (163950) | 33779033, 33683002 |
| 23 | F | 0.1 | FLNA | NM_001456.4 | c.7895G>T | p.(Ser2640Ile) | Het | LP | XL | Maternal | Heterotopia, periventricular, (300049) | 25686753 |
| 24 | F | 16.3 | SCARB2 | NM_005506.4 | c.994+1G>T | p.(?) | Hom | P | AR (novel) | Unknown | Epilepsy, progressive myoclonic 4, with or without renal failure (254900) | NA |
| 25 | F | 0.7 | SON | NM_138927.4 | c.1193del | p.(Pro398Leufs*2) | Het | P | AD (novel) | De novo | Zhu-Tokita-Takenouchi-Kim [ZTTK] syndrome (617140) | NA |
| 26 | F | 6.3 | NSD2 | NM_001042424.3 | c.2683C>T | p.(Pro895Ser) | Het | VUS | AD (novel) | Unknown | Rauch–Steindl syndrome (619695) | NA |
AD, autosomal dominant; AR, autosomal recessive; F, female; Hem, hemizygous; Hom, homozygous; Het, heterozygous; M, male; NA, not applicable; LP, likely pathogenic; P, pathogenic; PMID, PubMed identifier; Pt., patient; VUS, variant of uncertain significance; XL, X-linked.
b) insAGTTAGCGGAACGGCAGTGAATACGAATCACGGACTCATCTGCGGTGAACTCGACGGGTGCGTTCGGCAGGGAGCGGCAAATTTCTGCCAGGATCTTGCC.
- References
- 1. Moreno-De-Luca A, Myers SM, Challman TD, Moreno-De-Luca D, Evans DW, Ledbetter DH. Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet Neurol 2013;12:406–14.
[Article] [PubMed] [PMC]2. Gonzalez-Mantilla PJ, Hu Y, Myers SM, Finucane BM, Ledbetter DH, Martin CL, et al. Diagnostic yield of exome sequencing in cerebral palsy and implications for genetic testing guidelines: a systematic review and meta-analysis. JAMA Pediatr 2023;177:472–8.
[Article] [PubMed] [PMC]3. Srivastava S, Cohen JS, Vernon H, Barañano K, McClellan R, Jamal L, et al. Clinical whole exome sequencing in child neurology practice. Ann Neurol 2014;76:473–83.
[Article] [PubMed] [PMC]4. Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86:749–64.
[Article] [PubMed] [PMC]5. Pekeles H, Accogli A, Boudrahem-Addour N, Russell L, Parente F, Srour M. Diagnostic yield of intellectual disability gene panels. Pediatr Neurol 2019;92:32–6.
[Article] [PubMed]6. Han JY, Lee IG. Genetic tests by next-generation sequencing in children with developmental delay and/or intellectual disability. Clin Exp Pediatr 2020;63:195–202.
[Article] [PubMed] [PMC]7. Savatt JM, Myers SM. Genetic testing in neurodevelopmental disorders. Front Pediatr 2021;9:526779.
[Article] [PubMed] [PMC]8. Manickam K, McClain MR, Demmer LA, Biswas S, Kearney HM, Malinowski J, et al. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2021;23:2029–37.
[Article] [PubMed] [PMC]9. Wayhelova M, Vallova V, Broz P, Mikulasova A, Smetana J, Dynkova Filkova H, et al. Exome sequencing improves the molecular diagnostics of paediatric unexplained neurodevelopmental disorders. Orphanet J Rare Dis 2024;19:41.
[Article] [PubMed] [PMC]10. Choi J. Ending the diagnostic odyssey: making whole-exome/genome sequencing the first-line test for global developmental delay. Ann Child Neurol 2025;33:83–4.
[Article]11. Chung HJ, Yang D, Kim GH, Kim SK, Kim SW, Kim YK, et al. of the Korean Developmental Screening Test for Infants and Children (K-DST). Clin Exp Pediatr 2020;63:438–46.
[Article] [PubMed] [PMC]12. Jo YH, Choi SH, Yoo HW, Kwak MJ, Park KH, Kong J, et al. Clinical use of whole exome sequencing in children with developmental delay/intellectual disability. Pediatr Neonatol 2024;65:445–50.
[Article] [PubMed]13. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Washington, DC: American Psychiatric Publishing, 2013.14. Seo GH, Kim T, Choi IH, Park JY, Lee J, Kim S, et al. Diagnostic yield and clinical utility of whole exome sequencing using an automated variant prioritization system, EVIDENCE. Clin Genet 2020;98:562–70.
[PubMed] [PMC]15. Han H, Seo GH, Hyun SI, Kwon K, Ryu SW, Khang R, et al. Exome sequencing of 18,994 ethnically diverse patients with suspected rare Mendelian disorders. NPJ Genom Med 2025;10:6.
[Article] [PubMed] [PMC]16. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24.
[Article] [PubMed] [PMC]17. McCormick EM, Lott MT, Dulik MC, Shen L, Attimonelli M, Vitale O, et al. Specifications of the ACMG/AMP standards and guidelines for mitochondrial DNA variant interpretation. Hum Mutat 2020;41:2028–57.
[Article] [PubMed] [PMC]18. Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med 2020;22:245–57.
[Article] [PubMed] [PMC]19. Lee JY, Oh SH, Keum C, Lee BL, Chung WY. Clinical application of prospective whole-exome sequencing in the diagnosis of genetic disease: experience of a regional disease center in South Korea. Ann Hum Genet 2024;88:101–12.
[Article] [PubMed]20. Lee BL, Oh SH, Jun KR, Hur YJ, Lee JE, Keum C, et al. First Korean case of Coffin-Siris Syndrome with a novel frameshift ARID1B mutation. Ann Clin Lab Sci 2020;50:140–5.
[PubMed]21. Ahn JH, Oh SH, Park JK, Kim KH, Lee JE, Chung WY, et al. A novel frameshift CASK variant in a 6-month-old Korean female infant with global developmental delay, progressive microcephaly, and pontocerebellar hypoplasia: a case report. Ann Clin Lab Sci 2022;52:488–93.
[PubMed]22. Srivastava S, Love-Nichols JA, Dies KA, Ledbetter DH, Martin CL, Chung WK, et al. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med 2019;21:2413–21.
[Article] [PubMed] [PMC]23. Shin S, Lee J, Kim YG, Ha C, Park JH, Kim JW, et al. Genetic diagnosis of children with neurodevelopmental disorders using whole genome sequencing. Pediatr Neurol 2023;149:44–52.
[Article] [PubMed]