Pulmonary hemorrhage in pediatric lupus anticoagulant hypoprothrombinemia syndrome
Article information
Abstract
Lupus anticoagulant-hypoprothrombinemia syndrome (LAHPS), a very rare disease that is caused by the presence of antifactor II antibodies, is usually counterbalanced by the prothrombotic effect of lupus anticoagulant (LAC). Patients with LAHPS are treated using fresh frozen plasma, steroids, immunosuppressive agents, and immunoglobulins for managing the disease and controlling hemorrhages. Notably, steroids are the important treatment for treating hypoprothrombinemia and controlling the bleeding. However, some patients suffer from severe, life-threatening hemorrhages, when factor II levels remain very low in spite of treatment with steroids. Here, we report a case of LAHPS in a 15-year-old girl who experienced pulmonary hemorrhage with rapid progression. She was referred to our hospital owing to easy bruising and prolonged bleeding. She was diagnosed with LAHPS that presented with pancytopenia, positive antinuclear antibody, proloned prothrombin time, activated partial thromboplastin time, positive LAC antibody, and factor II deficiency. Her treatment included massive blood transfusion, high-dose methylprednisolone, vitamin K, and immunoglobulin. However, she died due to uncontrolled pulmonary hemorrhage.
Introduction
Lupus anticoagulant-hypoprothrombinemia syndrome (LAHPS) is a very rare disease associated with transient viral infections, drug reactions, antiphospholipid syndrome, systemic lupus erythematosus (SLE) and exists even in healthy individuals1,2,3). It is caused by antifactor II antibody, which is usually counterbalanced by the prothrombotic effect of lupus anticoagulant (LAC)1,2,3). Corticosteroid is required to control the hemorrhage and correct the hypoprothrombinemia, and is effective in many cases3,4). However, some cases with fatal course showed poor response to steroid treatment1).
We report a clinical case of a 15-year-old girl with SLE, who was diagnosed with LAPHS and died due to uncontrolled pulmonary hemorrhage caused by factor II deficiency in spite of supportive treatment including steroid and immunoglobulin.
Case report
A 15-year-old girl was admitted to the previous hospital due to easy bruising and prolonged bleeding after tooth extraction. She was easily fatigued and suffered from loss of appetite for several weeks. Laboratory data revealed pancytopenia. After several days, she was transferred to our hospital for further evaluation for pancytopenia. She had no past or family history of any bleeding disorders or connective tissue diseases.
The patient was stable in vital signs, and revealed no specific findings except for several bruises on both lower legs on physical examination. Initial laboratory tests showed a leukocyte count of 1,560/µL, hemoglobin 7.5 g/dL, and a platelet count 19,000 /µL. The patient's prothrombin time (PT) was 28.6 seconds (normal values, 10.1.14.0 seconds) and 23%, international normalized ratio was 2.65, and activated partial thromboplastin time (aPTT) was more than 120 seconds (normal values, 21.0.38.0 seconds). The disseminated intravascular coagulation (DIC) profile showed that 89% of antithrombin III (normal values, 80%.120%), 0.47 mg/L of d-dimer (normal values, <0.8 mg/L) and 1.08 µg/mL of fibrin degradation products (normal values, <5 µg/mL), and these results suggested that she was not complicated with DIC. The patient also had proteinuria (3+) and microscopic hematuria. There were no abnormalities on her chest radiography. The bone marrow biopsy, study for autoimmune disease and factor disease were planned to find out the cause of pancytopenia and prolongation of PT and aPTT. The patient was given blood transfusion with packed red cells, fresh frozen plasmas (FFPs), and single donor platelets.
On hospital day 2, the patient developed fever and cough. The chest radiography showed mild haziness with mild left pleural effusion, and she was started on intravenous antibiotics. Peripheral blood still showed pancytopenia and there was no improvement of prolongation of PT and aPTT in spite of blood transfusion including FFP.
On hospital day 4, an antibody screening test for cold antibody and the direct Coomb's test were positive. The complement levels were C3, 18 mg/dL; C4, <1.0 mg/dL; and CH50, <10 U/mL. Tests for antinuclear antibodies, antidouble stranded DNA antibodies, LAC antibodies, anticardiolipin antibodies immunoglobulin (Ig) M, SLE profile and syphilis reagin test were all positive. And antiphospholipid antibodies IgM/G and anticardiolipin antibodies IgG were negative. Repeated complete blood count, PT and aPTT showed no significant change in spite of continuous blood products transfusion. The bone marrow biopsy was normal. We diagnosed the patient with SLE based on revised criteria of American College of Rheumatology. The patient had no other symptoms except for purpura on lower extremeties, and radiographic finding showed no significant change compared to prior 2 days ago.
On hospital day 7, the patient complained of dyspnea. Moist rales were audible around all the lung fields. Follow-up x-rays showed slightly increased haziness on both lung fields. Evaluation of clotting factors revealed that the level of factor II was 3.3% (normal values, 50%-150%). We confirmed the patient with LAHPS. A 1:1 dilution of patient plasma with normal plasma partially corrected the PT and aPTT. Protein S activity was 147.4% (normal values, 65%-140%) and protein C activity, 76% (normal values, 70%-130%). We added high dose of intravenous methylprednisolone pulse therapy (20 mg/kg/day) but platelet count and prolongation of PT and aPTT did not improve. On hospital day 10, the dyspnea was aggravated due to the pulmonary hemorrhage and she was moved into the intensive care unit. Computed tomography scans for chest, abdomen and pelvis showed no thromboembolisms. Intravenous immunoglobulin (IVIG) treatment (300 mg/kg/day) had no effect for correction of PT and aPTT, but platelet count slightly increased. PT and aPTT were slightly improved after administration of IVIG for 2 days. The blood test results showed a platelet count of 78,000 /µL, PT of 42%, and aPTT of 88.5 seconds (Fig. 1). However, levels of aspartate aminotransferase, alanine aminotransferase, blood urea nitrogen and creatinine began to increase. We increased the dose of IVIG up to 1 g/kg/day, but her chest condition deteriorated very rapidly and multiorgan failure got worse, too. Finally she required intubation and mechanical respiratory support, and died due to aggravation of the pulmonary hemorrhage on hospital day 12.
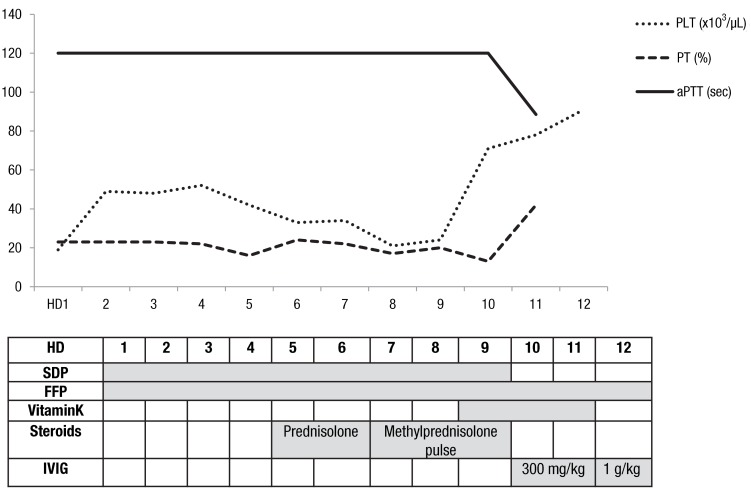
Laboratory findings of the patient and the treatment provided. HD, hospital day; PLT, platelet; PT, prothrombin time; aPTT, activated partial thromboplastin time; SDP, single donor platelet; FFP, fresh frozen plasma; IVIG, intravenous immunoglobulin.
Discussion
LAC is an antiphospholipid antibody associated with hypercoagulable states and thromboembolic events1,2,3). In most children of young age, the presence of LAC is transient, benign and postinfectious compared to autoimmune persisted type which mainly occurs in adolescence5). Bleeding is a rare manifestation of LAC, and when it occurs, it is nearly always due to thrombocytopenia or factor II deficiency2,6). Such bleeding results from the presence of antifactor II antibody, which usually counterbalances the prothrombotic effect of LAC1). And also, thrombosis was rare and strongly associated with persistently positive LAC5). LAC along with a specific nonphospholipid-dependent antiprothrombin antibody causes an acquired hypoprothrombinemia7).
LAPHS is a rare clinical disease. It occurs with viral infection and drug reactions in healthy individuals, or it appears mostly in young females with autoimmune disease like SLE1,2,3,6). Increasing incidence during adolescence and the gender distribution are similar to those of SLE5). Seventy-four cases of LAPHS has been reported worldwide according to a literature review retrieving the related articles published between 1960 and 2011. Epidemiologic characteristics of these 74 cases shows that 43 patients (58%) were aged <15 years at disease onset8). The mechanism suggested that antiphopholipid antibodies bound to prothrombin in circulation accelerate clearance rather than interfere with prothrombin function5). Some authors suggest cross-reactivity between the antiphopholipid antibodies and phopholipid epitopes in the prothrombin molecules1). The clinical findings are generally asymptomatic, but there can be severe hemorrhagic symptoms such as gynecologic bleeding, macroscopic hematuria, gastrointestinal bleeding, brain hematoma, diffuse muscular hematoma and venous or arterial thrombosis2,8). Despite the fact that about 50% of patients develop severe bleeding, the overall prognosis is quite good, with a mortality rate of less than 5%8).
Treatment of factor II deficiency should be individualized, and the underlying cause should be found and treated. Administration of FFP, prothrombin complex concentrates, vitamin K are useful. However, most of cases responded successfully to corticosteroid but not to vitamin K, FFP or blood transfusion3). Most of patients previously reported in the literature have responded well to prednisolone treatment alone3,4). A few cases required additional immunosuppressive drugs such as cyclophosphamide, azathioprine or rituximab because the patients had symptom recurrences when drug therapy was tapered, but immunosupression may not work immediately1,3,9). Therefore, corticosteroid therapy is the treatment of choice for LAHPS associated with SLE1,2,3,4,6,8). It decreases the clearance of the prothrombin antibody complexes and normalizes abnormal coagulation times, including those of PT, aPTT and clotting factors1,2,3,4,6). IVIG treatment also has been shown to be effective in the setting of acute bleeding1). In our patient, bleeding did not stop with steroid treatment. Coagulation studies improved significantly after administration of IVIG but the patient died because of uncontrolled pulmonary hemorrhage. We think that the administration of IVIG was started too late with an insufficient dose.
Our case shows that when bleeding occurs in SLE patients, the possibility of hypoprothrombinemia should be considered and prothrombin time and factor study should be measured3). Significant prolongation of the PT or aPTT requires further evaluation for autoantibodies against coagulation factors10). Management may require the use of intravenous steroids, immunoglobulin and immunosuppressive agents1,6,9). But if factor II levels are very low (under 10%), patients may experience severe life threatening bleeding such as uncontrolled pulmonary hemorrhage. LAHPS associated with autoimmune disease should be diagnosed and managed carefully. Early treatments are thought to be helpful because severe hemorrhagic complications are common. Moreover, the rapid progression of hemorrhage can be retarded by more aggressive treatments such as immunoglobulins and immunosuppressive agents.
Notes
No potential conflict of interest relevant to this article was reported.