A novel PRF1 gene mutation in a fatal neonate case with type 2 familial hemophagocytic lymphohistiocytosis
Article information
Abstract
Hemophagocytic lymphohistiocytosis (HLH) occurs in the primary form (genetic or familial) or secondary form (acquired). The familial form of HLH (FHL) is a potentially fatal autosomal recessive disorder that occurs because of constitutional defects in cell-mediated cytotoxicity. Here, we report a fatal neonatal case of type 2 FHL (FHL2) that involved a novel frameshift mutation. Clinically, the newborn presented with severe sepsis-like features and required mechanical ventilation and continuous venovenous hemodiafiltration. Flow cytometry analysis showed marked HLH and complete absence of intracytoplasmic perforin expression in cytotoxic cells; therefore, we performed molecular genetic analyses for PRF1 mutations, which showed that the patient had a compound heterozygous mutation in PRF1, that is, c.65delC (p.Pro22Argfs*2) and c.1090_1091delCT (p.Leu364Glufs*93). Clinical and genetic assessments for FHL are required for neonates with refractory fever and progressive multiple organ failure, particularly when there is no evidence of microbiological or metabolic cause.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory disorder with clinical manifestations that include fever, organomegaly, and cytopenia1). HLH occurs either as a primary form (genetic or familial) or as a secondary form (acquired)2). The familial form of HLH (FHL) is a potentially fatal autosomal recessive disorder due to constitutionally defective cell-mediated cytotoxicity3). In particular, the onset of FHL is reported to be within one year after birth in 70% to 80% of the cases. Until now, five genetic loci for FHL (FHL1-FHL5) have been reported, involving the following four causative genes: PRF1 (FHL2), UNC13D (FHL3), STX11 (FHL4), and STXBP2 (FHL5)3). In this report, we describe a fatal case of FHL2 due to a novel PRF1 gene mutation in a newborn girl with severe clinical manifestations that mimicked severe sepsis.
Case report
The proband was a 29-day-old female infant who was admitted to our institution because of four days of mild fever and rapid breathing, moaning, and mottling. She was the second child of a nonconsanguineous couple, born at 38+4 weeks of gestation with a birth weight of 3,500 g. Family history revealed that her sister died at 28 days of age from sepsis with rapidly progressive clinical manifestations of acute hepatitis, gastrointestinal bleeding, and pulmonary hemorrhage. The initial neonatal course of the proband was not remarkable until she developed respiratory distress and uncontrolled fever on postnatal day 29. She subsequently experienced significant bleeding and bruising from coagulopathy and had hepatosplenomegaly. Her condition deteriorated, and she developed lactic acidosis, 22.38 mmol/L (normal range, 0.5-2.2 mmol/L), which required mechanical ventilation and continuous veno-venous hemodiafiltration (Fig. 1).
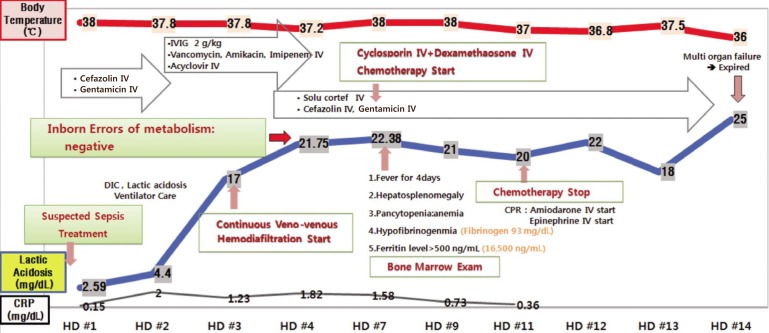
Hospital course and management of the patient. CRP, C-reactive protein (normal range, 0-0.3 mg/dL); IVIG, intravenous immunoglobulin; IV, intravenous; DIC, disseminated intravascular coagulation; CPR, cardiopulmonary resuscitation; HD, hospital day.
Leukopenia and anemia slowly developed along with thrombocytopenia. The 7th day of the admission, laboratory finding revealed leukocytes level at 4,010×103/µL, thrombocytes 28×103/µL, hemoglobin 9.7 g/dL, absolute neutropenic counts 960×103/µL, triglyceride 774 mg/dL, ferritin 165,000 ng/mL, fibrinogen 93 mg/dL.
Laboratory workup for infection revealed no diagnostic findings; however, on day 31, broad-spectrum antibiotics and intravenous immunoglobulin and corticosteroid were administered empirically with a presumptive diagnosis of neonatal sepsis. Since the patient's manifestations met the criteria for HLH 2004, a bone marrow study was performed on day 35, which revealed frequent active hemophagocytic histiocytes. As the first step of a diagnostic workup for FHL, we performed flow cytometric analysis for the intracytoplasmic perforin expression of cytotoxic cells using the proband's peripheral blood sample, as previously described with some modifications4). We observed a complete absence of perforin expression in the proband's natural killer (NK) cells and CD8+ T cells. The experiments using her parents' blood samples demonstrated partial deficiency of cytoplasmic perforin in NK cells and complete deficiency in CD8+ T cells (Fig. 2A). Based on the results of flow cytometric analysis, we obtained written informed consent from the parents and proceeded with a molecular genetic test for FHL2 by direct sequencing analysis of the PRF1 gene, as previously described5). As a result, the proband was shown to have two deleterious point mutations of PRF1, c.65delC [p.Pro22Argfs*2] and c.1090_1091delCT [p.Leu364Glufs*93] (Fig. 2B). And no other mutation has been found in direct sequencing analysis of the PRF1 gene. Her parents were heterozygous carriers of the respective mutant alleles (compound heterozygous for the c.65delC mutation (of maternal origin; novel) and the c.1090_1091delCT mutation (of paternal origin; known), confirming the compound heterozygous status of the two mutations in the proband. Based on the diagnosis of FHL2, the patient received HLH-2004 chemotherapy with cyclosporine, etoposide, and dexamethasone. She experienced hypotensive episodes that were initially responsive to fluid resuscitation and medications but that eventually progressed to refractory status, leading to renal failure. She died from multiorgan failure on day 43.
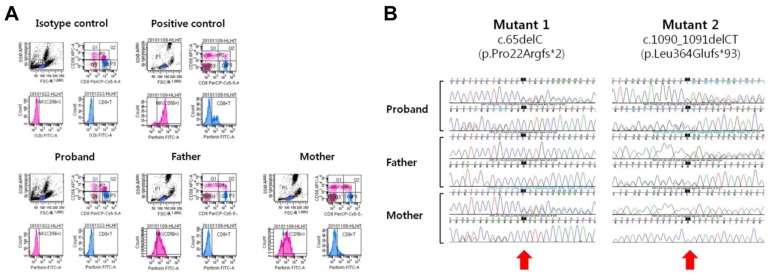
(A) Flow cytometry images for perforin protein expression in cytotoxic cells. The upper panel provides the results of an isotype and a normal control, and the lower panel provides the results of the proband and her parents. Note the complete deficiency of perforin expression both in the natural killer (NK) cells and in the CD8+ T cells in the proband. The carrier parents showed partial perforin deficiency in NK cells and complete deficiency in CD8+ T cells. (B) Direct sequencing analysis of the PRF1 gene showed that the proband was compound heterozygous for the c.65delC mutation (of maternal origin; novel) and the c.1090_1091delCT mutation (of paternal origin; known) (arrows).
Discussion
In this report, we described a fatal neonatal case of FHL2 due to mutations of PRF1, including a novel frameshift mutation. The patient's complicated multiorgan manifestations and fulminant clinical course mimicking severe sepsis delayed the diagnosis of FHL. The family history included a deceased sister with similar clinical manifestations, which led us to search for a genetic etiology. Since the onset of FHL is typically young, frequently <1 year6), clinical suspicion for FHL is critical for timely diagnosis and proper management to improve the prognosis of the disease in neonatal intensive care. Flow cytometric analysis of the intracytoplasmic perforin expression of cytotoxic cells and, more recently, intraplatelet expression of Munc 13-4 protein have been introduced to screen for FHL2 and FHL3, respectively4,7). Our patient showed a complete deficiency of perforin in both NK cells and CD8+ T cells, while her carrier parents had a partial deficiency in NK cells and a complete deficiency in CD8+ T cells, respectively. This observation is in line with the findings described in a previous report4).
At the molecular level, the patient was compound heterozygous for two point mutations, c.65delC (p.Pro22Argfs*2) and c.1090_1091delCT (p.Leu364Glufs*93). Both mutations are deleterious in nature by way of introduction of a premature stop codon due to a frameshift. The complete deficiency of perforin expression revealed by flow cytometric analysis could be interpreted in the context of a genotype-phenotype correlation: two deleterious mutant alleles resulting in severe protein deficiency and the early age of onset followed by a fulminant clinical course.
Based on a review of the literature and mutation database (Human Gene Mutation Database Professional; http://www.hgmd.org/), c.65delC is a novel mutation. The c.1090_1091delCT mutation had been reported in a Korean patient (a two-month-old girl) with homozygous status in our previous study5). Thus, it could be a recurrent mutation of FHL2 in Korea. Interestingly, c.1090_1091delCT was reported to be the most frequent FHL2 mutation in Japan8). The small number of FHL2 in Korea is due to the unusual predominance of FHL3 (~90%)5). Collectively, c.1090_1091delCT is restricted to FHL2 of Korean and Japanese descendents with high frequencies, suggesting a distinct geographical distribution and founder effect, as exemplified by PRF1:c.50delT mutation in FHL2 in people of African ancestry9).
Clinically, our patient had a course that rapidly deteriorated into multiorgan failure requiring continuous veno-venous hemodiafiltration. Severe lactic acidosis and a family history of a sibling who had died from similar clinical manifestations that mimicked sepsis led us to suspect bacterial sepsis with a genetic etiology of metabolic disease. The suspicion of FHL was delayed until we obtained negative results for bacteriologic and metabolic workup. However, persistent fever, sepsis-like features, and a progressively deteriorating clinical course may suggest the diagnosis of FHL in neonates10). In previous data, three cases of FHL in identical male twins and their younger brother who presented with fever and severe hepatosplenomegaly had been reported. One of them showed reversed CD4/CD8 ratio (0.52) in flowcytometric lymphocyte subset analysis and aspirate of bone marrow revealed typical features consistent with FHL in two of them. But none of them performed direct sequencing analysis11). As exemplified by the present case, FHL needs to be considered as one of the differential diagnoses when a newborn presents with severe sepsis-like features with progressive multiorgan failure.
In summary, we described a fatal neonatal case of FHL2 that mimicked severe sepsis. Clinical assessment and laboratory workup for FHL is needed in critically ill neonates with severe sepsis and multiorgan failure, particularly when there is no evidence of a microbiological or metabolic cause.
Acknowledgments
This study was supported by a grant from the Clinical Research Development Program at Samsung Medical Center, Seoul, Korea (#CRS111-03-1).
Notes
No potential conflict of interest relevant to this article was reported.