A case of isodicentric chromosome 15 presented with epilepsy and developmental delay
Article information
Abstract
We report a case of isodicentric chromosome 15 (idic(15) chromosome), the presence of which resulted in uncontrolled seizures, including epileptic spasms, tonic seizures, and global developmental delay. A 10-month-old female infant was referred to our pediatric neurology clinic because of uncontrolled seizures and global developmental delay. She had generalized tonic-clonic seizures since 7 months of age. At referral, she could not control her head and presented with generalized hypotonia. Her brain magnetic resonance imaging scans and metabolic evaluation results were normal. Routine karyotyping indicated the presence of a supernumerary marker chromosome of unknown origin (47, XX +mar). An array-comparative genomic hybridization (CGH) analysis revealed amplification from 15q11.1 to 15q13.1. Subsequent fluorescence in situ hybridization analysis confirmed a idic(15) chromosome. Array-CGH analysis has the advantage in determining the unknown origin of a supernumerary marker chromosome, and could be a useful method for the genetic diagnosis of epilepsy syndromes associated with various chromosomal aberrations.
Introduction
Supernumerary marker chromosomes (SMCs) are relatively common in the field of clinical cytogenetics, and their prevalence is estimated to be 0.05% of unselected live-born infants1). It is estimated that 50% of all SMC are derived from chromosome 15, and that approximately 80% of them represent an isodicentric chromosome 15 (idic(15) chromosome)2). A high prevalence of idic(15) chromosome, out of the supernumerary marker chromosomes, may favor fluorescence in situ hybridization (FISH) that targets the region of idic(15) as the next diagnostic procedure. However, array comparative genomic hybridization (CGH) could provide additional advantages by searching the whole chromosomal region, and evaluating the extent of the region in the diagnosis of patients with SMCs. Recent studies using an array CGH to investigate patients with intellectual disabilities (with or without dysmorphic features) have demonstrated the ability to improve the diagnostic detection rate of small chromosomal abnormalities3-5). We report a new patient with an idic(15) chromosome, presented with severe developmental delay, hypotonia, and epilepsy, and was diagnosed with molecular techniques, such as array CGH and FISH, to further define the involved regions.
Case report
A 10-month-old female infant was referred to our pediatric neurology clinic for uncontrolled seizures and global developmental delay. She was born at term with normal birth weight (2,900 g) after a normal pregnancy and delivery. Her mother did not take any medications during pregnancy. At birth, her mother was 38 years old and her father was 39. Her older brother was healthy. There were no other family members with neurodevelopmental disorders. At the time of the referral, she could not control her head nor follow moving objects. The head circumference, height, and weight were all below the 3rd percentile. A physical examination was rather unspecific, and might include: minor facial dysmorphisms, such as frontal bossing, wide-set eyebrows, down-slanting palpebral fissures, low-set ears, highly arched palate, broad nose, short philtrum, strabismus, and generalized hypotonia. A metabolic evaluation (that included urine organic acids, serum amino acids, and a thyroid function tests) was normal. A brain magnetic resonance imaging showed mild brain atrophy. She suffered from generalized tonicclonic or generalized tonic seizures from the age of 7 months. Video-electroencephalography (EEG) monitoring was conducted at the age of 11 months and 25 months. At the first monitoring, brief tonic extension of both arms and legs were accompanied by ictal EEG pattern which showed mild diffuse attenuation of background activities with low amplitude rhythmic theta activities. At the second monitoring, clusters of extensor spasms were accompanied by ictal EEG which showed diffuse delta bursts followed by electrodecremental response. Video-EEG monitoring results were consistent with generalized tonic seizures and epileptic spasms. At the age of 6 years, she has been seizure free for 3 years after receiving valproate, topiramate, and lamotrigine. However, her developmental status has not improved and she still presents hand stereotypy, bruxism, and episodic tonic posturing.
Routine karyotyping revealed the presence of a supernumerary marker chromosome of the unknown origin (47, XX, +mar) (Fig. 1). Her parent's karyotypes were normal. Array comparative genomic hybridizations were performed using a Human Genome CGH Microarray Kit 180K, using an Agilent Oligo Array-CGH Hybridization Kit (Agilent Technologies, Palo Alto, CA, USA). Images of the arrays were acquired using a high-resolution microarray scanner (Cat. no. G2565CA, 2008, Agilent Technologies), and image analysis was performed using the genomic workbench standard edition 5.0 (ver. 5.0.14, 2009, Agilent Technologies). The result revealed amplification from 15q11.1 to 15q13.1 spanning 8.47 Mb (Fig. 2). Subsequent FISH analysis confirmed that the amplified chromosomal region, detected by an array CGH, was localized to the supernumerary marker chromosome (Fig. 3).
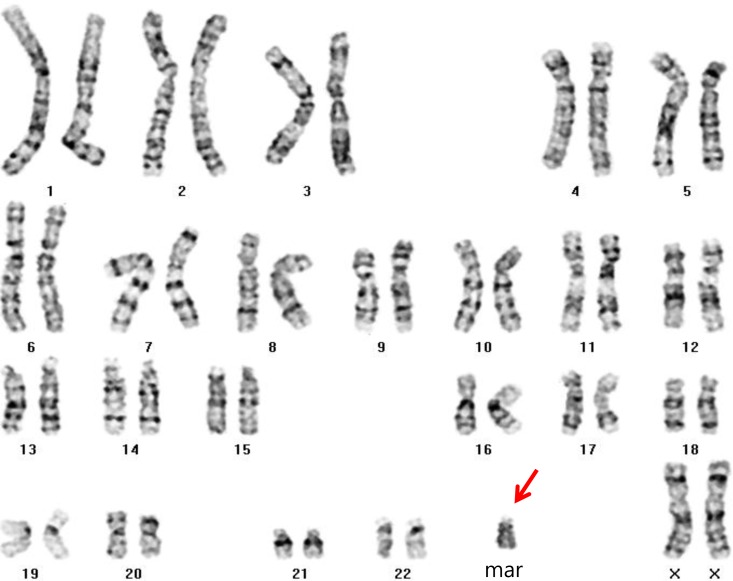
Routine karyotyping (GTG banding, ×1,000) showing the presence of a supernumerary marker chromosome of unknown origin (47, XX, +mar; arrow).
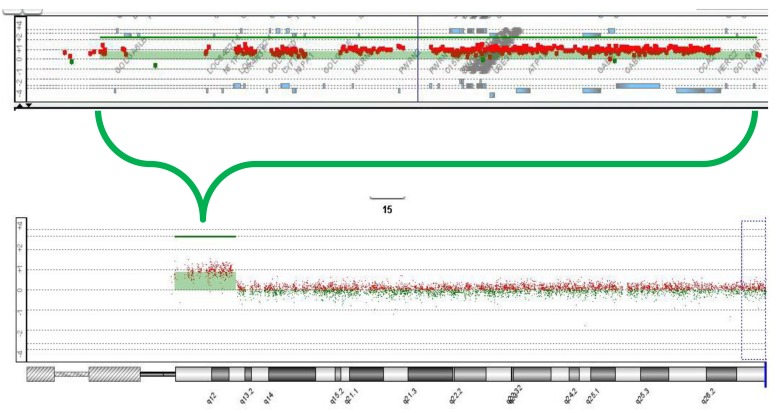
Array-comparative genomic hybridization analysis (Agilent, 180K oligonucleotide array) results showing amplification from 15q11.1 to 15q13.1, spanning 8.47 Mb.
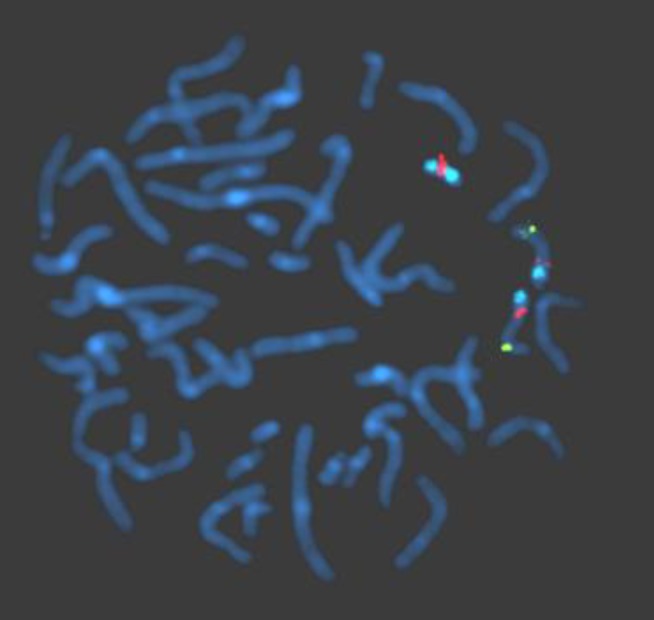
Fluorescence in situ hybridization analysis performed using probes, SNRPN, PML, and CEP 15. The supernumerary marker chromosome showing 1 SNRPN signal (orange) in the 15q11-q13 region and 2 distinct CEP 15 signals (aqua) in the 15p11.2 region. The hybridization signals observed on the distal long arm of chromosome 15 representing the control probe (PML, green), previously mapped to 15q22.
Discussion
The idic(15) syndrome displays clinical findings represented by early central hypotonia, developmental delay, intellectual disability, epilepsy, and autistic behavior6). Although minor facial dysmorphisms, such as down-slanting palpebral fissures, epicanthal folds, deep-set eyes, low-set ears, and highly arched palate, have been described, and other major malformations have not been frequently associated. Thus, epilepsy may be one of the principal symptoms that require therapeutic interventions from early infancy, and provide a diagnostic clue.
Many case reports7-10) and case series11-13) suggested that there is a predominance of generalized epilepsy, resembling infantile spasms, Lennox-Gastaut syndrome, with tonic, atonic, atypical absences, myoclonic, and epileptic spasms. Our case showed epileptic spasms and generalized tonic seizures, without generalized slow spike wave complexes, that could be classified as unspecified generalized epilepsy syndrome. The clinical presentations of the present case consist of generalized seizures and developmental delay that might be nonspecific enough to suspect chromosomal aberration syndrome and the chromosomal analysis performed a crucial role in the genetic diagnosis of the case. Therefore, we suggest that chromosomal analysis should be included in the diagnostic evaluation of patients with epilepsy and developmental delay, when an evaluation for structural brain lesions and metabolic disorders reveal no abnormal findings.
Limited reports have analyzed the extent of the amplified region in idic(15) syndrome patients14,15). Subsequently, genotype-phenotype correlations have not been systematically investigated. Genotype-phenotype correlations could be aided by detailed information of the involved genes in the amplified region, since the array CGH technique could localize the supernumerary marker chromosome (other than chromosome 15) as well as estimate the extent of the amplified region. The previous study by Wang et al.14) analyzed the breakpoint of idic(15) chromosomes, using array CGH. Two types of the distal breakpoints were demonstrated: one was BP3, and the other was BP5, including asymmetric amplification between BP4 and BP5. Array CGH analysis of the present case revealed the amplification of chromosome 15, ranging from 18,824,422 to 26,830,931 (National Center for Biotechnology Information build 36), corresponding to the distal breakpoint of BP3. The amplified region encompasses gamma-aminobutyric acid receptor subunit genes: α5 and α3. Alterations of these subunit genes have been involved with human childhood absence epilepsy16), and animal models of epilepsy characterized by seizures, jerky gait, and ataxia17). Interestingly, cases with 15q13.3 microdeletion and duplication, which corresponds to a region between BP4 and BP5, are increasingly reported18,19). The reported clinical features of patients with 15q13.3 microdeletion and duplication are rather heterogeneous, ranging from mental retardation and psychiatric illness to seizures. CHRNA7 that encode alpha 7 subunit of nicotinic acetylcholine receptors was specifically postulated to be involved with various neuropsychiatric symptoms, among the several genes located within this region; however, its pathologic significance remains to be further clarified20). Nevertheless, it would be interesting whether the differences in size of the amplified region in idic(15) cases would be associated with different clinical phenotypes. For this purpose, array CGH could have a clear advantage over other molecular techniques and contribute to broadening of the phenotype, even if the chromosomal aberrations were previously described.
Acknowledgment
This study was supported by a grant from the Life Insurance Philanthropy Foundation.