Recent updates on systemic treatment of atopic dermatitis
Article information

Abstract
Atopic dermatitis (AD) is a complex disease with multifactorial pathogenesis and variable clinical presentation. Up to one-fifth of patients with AD develop moderate to severe disease that is often refractory to classical therapies and can compromise quality of life. This review summarizes recent clinical evidence on biological agents and small-molecule immunotherapies for the treatment of AD.
Key message
This review details how to best apply biological agents and small-molecule immunotherapies for the treatment of atopic dermatitis in clinical practice.
Introduction
Atopic dermatitis (AD) is a chronic relapsing inflammatory cutaneous disorder characterized by eczematous skin lesions and pruritus [1,2]. AD is among the most commonly occurring inflammatory dermatological conditions, affecting an estimated 14% of children and 1%–3% of adults in Korea [3-5].
The pathogenesis of AD is multifaceted, involving a combination of functional defects of skin barriers, genetic components, and immunological dysfunction. It is now known that the immune response universally seen in most patients with AD is skewed toward T helper lymphocyte type 2 (Th2) cytokines, while other T helper cell pathways, including Th1, Th17, and Th22, show more heterogenous activation patterns [6]. A better understanding of this complex immunological milieu has led to the recent development of several novel therapies, including biologic agents that inhibit Th2 cytokines, small-molecule inhibitors that hinder downstream inflammatory signaling pathways, and several other candidate molecules. Such robust progress has led many patients who were previously disappointed by ineffective treatment with, or side effects caused by, traditional therapies such as emollients, local topical therapies, and systemic immunotherapies. However, a clear guideline on the clinical application of these novel agents has yet to be established.
Here we review the emerging evidence of the treatment of AD with focus on biologics and small-molecule inhibitors and attempt to shed light on how best to apply them in clinical practice.
Biologics
1. Dupilumab
Dupilumab is a human monoclonal antibody targeting interleukin-4 receptor alpha (IL4R ) that inhibits interleukin-4 (IL4) and interleukin-13 (IL13), Th2 cytokines that play an important role in the pathogenesis of AD. Dupilumab was first approved by the U.S. Food and Drug Administration (FDA) in 2017 for adults with moderate to severe AD that was not adequately controlled by topical therapies. The indication was subsequently expanded in 2020 to include adolescents aged 12–17 years in 2019 and children aged 6–12 years. Most recently, in 2022, dupilumab was approved for the treatment of moderate to severe AD in younger children aged 6 months to 5 years. In Korea, dupilumab is currently approved for the treatment of moderate to severe AD in adults >18 years of age, adolescents 12–17 years of age, and children 6 months to 11 years of age.
The efficacy and safety of dupilumab in adults with AD were evaluated in the SOLO1 and SOLO2 randomized, placebo-controlled, phase 3 trials of identical design that evaluated 671 patients with moderate to severe AD that was inadequately controlled by topical treatment. Patients were randomly assigned at a 1:1:1 ratio to receive, for 16 weeks, weekly subcutaneous injections of dupilumab 300 mg or placebo or the same dose of dupilumab every 2 weeks (Q2W) alternating with placebo. The proportion of patients with an Investigator's Global Assessment (IGA) score of 0 or 1 after 16 weeks of treatment was significantly higher in each of the 2 dupilumab groups compared to the placebo group, with 38% of the patients receiving dupilumab Q2W and 37% of those receiving weekly dupilumab achieving the said endpoint compared to 10% of those receiving placebo (P<0.001 for both comparisons with placebo). Similarly, an improvement of at least 75% on the Eczema Area and Severity Index (EASI-75) at week 16 was reported in significantly more patients receiving each dupilumab regimen than among those receiving placebo (P<0.001 for all comparisons) [7].
The phase 3 LIBERTY AD ADOL trial evaluated 251 adolescents aged 12–17 years with inadequately controlled moderate to severe AD. The patients were randomly assigned to receive dupilumab 200 mg or 300 mg Q2W, dupilumab 300 mg every 4 weeks (Q4W), or placebo. The dupilumab groups showed higher proportions of patients with EASI75 improvement (Q2W, 41.5%; Q4W, 38.1%; placebo, 8.2%) and a higher proportion of patients reaching an IGA score of 0 or 1 (Q2W, 24.4%; Q4W, 17.9%; placebo, 2.4%), and both regimens showed significant improvement over placebo (P<0.001 in all cases) [8].
In the LIBERTY AD PEDS trial, 367 children aged 6–11 years were assigned in 1:1:1 ratio to receive dupilumab 300 mg Q4W, dupilumab 100 mg or 200 mg Q2W, or placebo. At week 16, significantly higher number of patients receiving either dupilumab regimen achieved the coprimary endpoints: a significantly greater proportion of patients achieved an IGA score of 0 or 1 and EASI-75 improvement compared to placebo [9]. Similarly, the LIBERTY AD PRESCHOOL trial targeting 162 children aged 6 months to 5 years reported a significantly greater proportion of patients reaching an IGA score of 0 or 1 and EASI-75 improvement with dupilumab versus placebo [10].
Common adverse events (AEs) reported across trials included AD exacerbation and infections such as conjunctivitis, blepharitis, nasopharyngitis, and upper respiratory tract infections, but they were rarely severe and infrequently led to treatment discontinuation [7-10].
Some real-world data suggest that, despite the generally effective and well-tolerated nature of dupilumab, it may not be as effective in the real world as in clinical trials due to the operator-dependency of the outcomes used to evaluate responses (e.g., to IGA and EASI-75) [11]. However, a real-world Korean study of 101 patients demonstrated a similar efficacy and safety profile to those reported by clinicaltrials. The same study also found that a persistent lactate dehydrogenase elevation and hypereosinophilia were negatively correlated with a treatment response to dupilumab. These findings were maintained in the subsequent 52-week follow-up [12,13]. Other Korean data have also demonstrated real-world efficacy and safety similar to those of pivotal trials, although the predictive values of certain laboratory markers require further exploration [14,15].
2. Tralokinumab
Tralokinumab is a fully human immunoglobulin G4 monoclonal antibody that binds with high affinity to IL13 and inhibits its interaction with IL13R. It neutralizes the biological activity of IL13 by blocking its interaction with the IL13Rα1/IL4Rα, which greatly decreases Th2 inflammatory mediators [13]. Tralokinumab was first approved by the FDA in 2022 for adults with moderate to severe AD. This indication was expanded in 2023 to include adolescents aged 12–17 years. Also in 2023, tralokinumab obtained regulatory approval in Korea for adults and adolescents with moderate to severe AD.
The ECZTRA 1 and ECZTRA 2 randomized, double-blind, multicenter, placebo-controlled phase 3 trials evaluated the safety and efficacy of tralokinumab in adults. The patients were randomized 3:1 to receive tralokinumab 300 mg or placebo Q2W. At week 16, those who achieved an IGA of 0 or 1 or EASI-75 improvement were again randomized to tralokinumab 300 mg, tralokinumab 300 mg, or placebo Q2W. Tralokinumab 300 mg Q2W resulted in a significantly higher proportions of patients who achieved an IGA of 0 or 1 (15.8% vs. 7.1% in ECZTRA 1 [95% confidence interval {CI}, 4.1–13.1; P=0.002] and 22.2% vs. 10.9% in ECZTRA 2 [95% CI, 5.8–16.4; P<0.001]) and EASI-75 improvement (25.0% vs. 12.7% [95% CI, 6.5–17.7; P<0.001] and 33.2% vs. 11.4% [95% CI, 15.8–27.3; P<0.001]) by week 16 [16].
Combination therapy with tralokinumab and topical corticosteroids (TCS) were evaluated in 2 randomized, double-blind, placebo-controlled studies of adults with moderate to severe AD (ECZTRA 3) and adults with severe AD and inadequate responses to or intolerance of ciclosporin A (ECZTRA 7). Both studies reported significantly greater proportions of patients achieving EASI-75 improvement by week 16. The ECZTRA 3 trial further showed statistically significant improvement in all key secondary endpoints including SCORing Atopic Dermatitis scores and the proportion of patients achieving a ≥4-point reduction in weekly average daily Worst Pruritus Numerical Rating Scale (WPNRS) score [17,18].
A randomized double-blind placebo-controlled phase 3 trial (ECZTRA 6) assessed the efficacy and safety of tralokinumab in adolescents aged 12–17 with moderate to severe AD. The patients were randomized to receive tralokinumab 150 mg, tralokinumab 300 mg, or placebo Q2W for 16 weeks. The study reported significantly higher proportions of patients achieving an IGA score of 0 or 1 at week 16 with tralokinumab 150 mg (21.4%) and tralokinumab 300 mg (17.5%) compared to placebo (4.3%) (adjusted difference, 17.5% [95% CI, 8.4%–26.6%], P<0.001 for tralokinumab 150 mg; and 13.8% [95% CI, 5.3%–22.3%], P=0.002 for tralokinumab 300 mg vs. placebo). Significantly higher proportions of patients also achieved EASI-75 improvement by week 16 with tralokinumab 150 mg (28.6%) and tralokinumab 300 mg (27.8%) versus placebo (6.4%) (adjusted difference, 22.5% [95% CI, 12.4–32.6%], P<0.001 for tralokinumab 150 mg; and 22.0% [95% CI, 12.0%–32.0%], P<0.001 for tralokinumab 300 mg vs. placebo) [19].
A pooled safety analysis of 5 randomized double-blind placebo-controlled phase 2 and 3 trials of tralokinumab in adults with moderate to severe AD showed a safety and AE profile that was well-tolerated and consistent. The most common AEs with tralokinumab were upper respiratory tract infection, conjunctivitis, and injection-site reaction [20].
3. Lebrikizumab
Lebrikizumab is a selective high-affinity immunoglobulin G4 monoclonal antibody that targets soluble interleukin-13 (IL13) and prevents IL4R -IL13R 1 heterodimer formation. Due to this mechanism of action, lebrikizumab inhibits IL13 signaling without affecting the IL4 signaling pathway.
A phase 2b double-blind placebo-controlled randomized clinical trial evaluated lebrikizumab efficacy in 280 patients with moderate to severe AD treated for up to 16 weeks. The patients were randomized to receive lebrikizumab 125 mg Q4W, 250 mg Q4W, or 250 mg Q2W or placebo. At the end of the study, the lebrikizumab-treated groups showed a dose-dependent statistically significant improvement in EASI score compared with placebo. Similar improvements were seen for pruritus Numerical Rating Scale (NRS) score. Other key secondary endpoints, including IGA 0/1 response, EASI-50 improvement, EASI-75 improvement, and lesion severity, also showed dose-dependent improvement with lebrikizumab versus placebo [21].
The ADvocate1 and ADvocate2 were identically designed phase 3 trials that evaluated the efficacy and safety of lebrikizumab monotherapy in adolescents and adults with moderate to severe AD. At the end of week 16, IGA 0/1 was achieved by 43.1% and 33.2% of patients in the ADvocate 1 and ADvocate 2 trials, respectively, versus 10.8%–12.7% in the placebo group. EASI-75 improvement was achieved in 58.8% and 52.1% of patients receiving lebrikizumab in the ADvocate 1 and ADvocate 2 trials, respectively, compared with 16.2%–18.1% of patients receiving placebo. Higher proportions of patients receiving lebrikizumab also showed at least 4-point improvement in pruritus NRS scores compared to placebo (45.9% and 39.8% in the ADvocate 1 and ADvocate 2 trials, respectively, compared with 11.5%–13% for placebo). Lebrikizumab also showed significant improvement over placebo in other key secondary outcomes, including 90% improvement in EASI at week 16, a 4-point or greater improvement in pruritus NRS score at weeks 4 and 16, a 2-point or greater reduction in Sleep-Loss Scale score by week 16, and an IGA response at week 4 (P<0.05 for all vs. placebo). The only secondary outcome that did not reach statistical significance was a 4-point or greater reduction in pruritus NRS score by week 2 in the ADvocate 2 trial [22].
Lebrikizumab is not yet approved for use in Korea, and there are no Korean or other real-world data available on lebrikizumab to date (Table 1).

Summary of results of pivotal trials of biologics for treating AD
JAK Inhibitors
1. Upadacitinib
Upadacitinib is a small-molecule Janus kinase (JAK) inhibitor with a greater inhibitory potency for JAK1 than JAK2/3 and tyrosine kinase 2. Upadacitinib obtained FDA approval in 2022 based on 3 phase 3 pivotal trials, which are reviewed in detail below. In Korea, it is approved for use in adults and adolescents with moderate to severe AD.
Measure Up 1 and Measure Up 2 are replicate multicenter, randomized, double-blind, placebo-controlled phase 3 trials that evaluated 847 adolescents and adults with moderate to severe AD. The patients were randomly assigned to upadacitinib 15 mg once daily (OD), upadacitinib 30 mg OD, or placebo. Coprimary efficacy endpoints were met at the end of week 16, with significantly greater proportions of patients reaching EASI-75 improvement and a validated Investigator’s Global Assessment of AD (vIGA-AD) score of 0 or 1 with both upadacitinib dosage forms compared to placebo. In the Measure Up 1 trial, 70% and 80% of patients in the upadacitinib 15 mg and 30 mg groups, respectively, showed 75% improvement in EASI by week 16 versus only 16% of the placebo group. Similarly, 48% and 62% of the upadacitinib 15 mg and 30 mg groups, respectively, achieved a vIGA-AD score of 0 or 1 versus 8% of the placebo group. The Measure Up 2 trial showed similar patterns of improvement with an adjusted difference in the EASI-75 response of 46.9% (upadacitinib 15 mg vs. placebo) and 59.6% (upadacitinib 30 mg vs. placebo) and an adjusted difference in the vIGA-AD response of 39% (upadacitinib 15 mg vs. placebo) and 52% (upadacitinib 30 mg vs. placebo) [23].
A randomized double-blind placebo-controlled phase 3 trial, the AD Up, evaluated the efficacy and safety of upadacitinib plus TCS in 901 adults and adolescents with moderate to severe AD. As early as week 2 of treatment, both upadacitinib cohorts showed significantly higher proportions of patients who achieved EASI-75 improvement and a vIGA-AD score of 0 or 1 compared to placebo. This effect was maintained until week 16 of treatment (P<0.0001 for all cases). Improvements in itch NRS scores were also seen as early as week 1 of treatment, with both upadacitinib groups showing significantly greater proportions of patients who achieved at least 4-point improvement in WP-NRS scores [24].
The efficacy and safety of upadacitinib versus dupilumab were compared in adults with moderate to severe AD in the 24-week, head-to-head, randomized, double-blind, double-dummy phase 3b Heads Up trial. The patients were randomized 1:1 to receive upadacitinib 30 mg OD or dupilumab 300 mg Q2W. At week 16, a significantly greater proportion of patients receiving upadacitinib versus dupilumab achieved EASI-75 improvement (71% vs. 61.1%, respectively; 95% CI, 2.9%–17.0%; P=0.006), thus meeting the primary efficacy endpoint. This effect was observed as early as week 2 of treatment, with 43.7% of patients on upadacitinib versus 17.4% on dupilumab achieving EASI-75 improvement (P<0.001). The percentage improvement from baseline of WPNRS scores was also significantly greater in the upadacitinib group at week 16 (66.9% vs. 49.0%, P< 0.001) [25].
Safety analyses suggested that the incidence of treatment-emergent AEs (TEAEs) was greater with upadacitinib than placebo, while the incidence of serious AEs leading to discontinuation remained similar across treatment groups. Common TEAEs include acne, upper respiratory tract infection, and nasopharyngitis [24].
A real-world prospective study of 146 adult patients showed that the upadacitinib 15 mg and 30 mg dosage forms effectively controlled the disease with no statistically significant intergroup differences. Dose alterations occurred quite frequently, with 33 treatment courses of dose reduction and 15 courses of dose escalation in a total of 38 patients. Despite the flexible dosing regimen, the efficacy was well maintained with comparable results to the pivotal trials [26].
2. Baricitinib
Baricitinib, an oral selective JAK1 and JAK2 inhibitor, was granted European Medicines Agency regulatory approval in 2020 and Korean regulatory approval in 2021. FDA approval of baricitinib for AD remains pending.
BREEZE-AD1 and BREEZE-AD2, independent 16-week randomized, double-blind, parallel-group, placebo-controlled phase 3 trials, evaluated 1,239 adult patients with moderate to severe AD. The patients were randomized to receive baricitinib 1 mg, 2 mg, or 4 mg or placebo OD. The primary outcome was the superiority of baricitinib 2 mg or 4 mg compared to placebo as assessed by the proportion of patients reaching a vIGA-AD score of 0 or 1 by week 16. At week 16, all baricitinib groups in the BREEZE-AD1 trial achieved statistically significant improvement in vIGA-AD scores compared to placebo, with 11.8%, 11.4%, and 16.8% for the baricitinib 1 mg, 2 mg, and 4 mg groups, respectively, compared to 4.8% for placebo (P≤0.05 for 1 mg and 2 mg, P≤0.001 for 4 mg). In the BREEZE-AD2 trial, 8.8%, 10.6%, and 13.8% of patients receiving baricitinib 1 mg, 2 mg, and 4 mg achieved a vIGA-AD score of 0 or 1 compared to 4.5% receiving placebo (P≤0.05 for 2 mg, P≤0.001 for 4 mg) [27].
Combination therapy with baricitinib and TCS was assessed in another multicenter, double-blind, placebo-controlled, parallel-arm, phase 3 trial, BREEZE-AD 7. Patients were randomly assigned to receive placebo or baricitinib 2 mg or 4 mg OD. At week 16, a statistically significant vIGAAD benefit was seen with baricitinib 4 mg versus placebo, whereas baricitinib 2 mg failed to demonstrate a significant benefit. Baricitinib 4 mg treatment also showed significant improvement in the proportion of patients achieving EASI75 improvement at week 16, proportion of patients achieving at least a 4-point improvement in itch NRS scores at weeks 4 and 16, and mean change from baseline in skin pain NRS scores [28].
The incidence of TEAEs was similar across treatment groups in the BREEZE-AD 1 and BREEZE-AD2 trials but higher with baricitinib than placebo in the BREEZE-AD7 trial. Nasopharyngitis, upper respiratory tract infections, headaches, oral herpes, and acne were among the most common AEs seen in the 3 trials. Moreover, the incidence of serious AEs was similar across treatment groups in all 3 trials [27,28].
3. Abrocitinib
Abrocitinib, an oral JAK1 selective inhibitor, is approved for use by the FDA in adults and adolescents with moderate to severe AD. JADE MONO 1 and JADE MONO 2 are 2 identically designed, multicenter, international, placebo-controlled, parallel-group phase 3 trials that evaluated the efficacy and safety of abrocitinib monotherapy in adults and adolescents with moderate to severe AD. Patients received abrocitinib 100 mg or 200 mg or placebo OD for 12 weeks. Both abrocitinib groups achieved the primary endpoint of an IGA response benefit (i.e., proportions of patients achieving an IGA of 0 or 1 at week 12) and EASI-75 score improvement (i.e., proportions of patients achieving EASI-75 improvement at week 12) compared to the placebo group [29,30].
JADE TEEN, another phase 3 trial that assessed the efficacy and safety of abrocitinib plus TCS in adolescents aged 12–17 with moderate to severe AD showed similar IGA and EASI-75 response benefits, with the abrocitinib 100 mg and 200 mg groups showing significantly greater proportions of patients achieving the desired outcome at week 12 [31].
A more recent randomized double-blind active-controlled phase 3 trial compared the efficacy and safety of abrocitinib and dupilumab. Patients were randomized to receive abrocitinib 200 mg OD or dupilumab 300 mg Q2W. The abrocitinib group showed significantly higher proportions of patients who achieved at least 4-point improvement in Peak Pruritus NRS scores and those who achieved EASI-90 improvement at week 4 than the dupilumab group (Table 2) [32].

Summary of results of pivotal trials of Janus kinase inhibitors for treating AD
Discussion
With the emergence of novel systemic therapies for AD, it is more important than ever for the physician to first correctly identify a suitable candidate for systemic therapy and then create an individually tailored regimen for each patient.
Diagnosis
Apart from utilizing established diagnostic criteria to assess clinical and histopathological findings, itis important to rule out any condition that may present similarly to AD. In infants, several pathologies, including seborrheic dermatitis, nutritional deficiencies, metabolic disorders, and immunological disorders, can be mistaken for AD. In adults, other forms of dermatitis, such as contact dermatitis or nummular dermatitis, as well as cutaneous T-cell lymphoma can confound the diagnosis [33].
Patient identification and treatment decision-making
Traditional means of treatment for moderate to severe pediatric AD have been quite limited to date. The long-term use of systemic steroids is not recommended, while long-term safety data on pediatric use of systemic immunotherapies are lacking. Fortunately, with the recent progress in our understanding of the pathophysiology of AD and the resulting proliferation of novel therapies, physicians now have to luxury of being able to choose a treatment option based on drug efficacy, onset of action, mode of administration, and AEs as well as patient age and comorbidities.
Considering the significant impact that AD exerts on patient quality of life as well as the subjective nature of the perception of disease control, a shared decision-making process between the physician and the patient is crucial to optimizing the results. A previous retrospective study of Korean patients with AD demonstrated that EASI is not correlated with patient quality of life, while subjective measurement tools such as the Patient-Oriented Eczema Measure and NRS scores show strong correlations [34]. Accordingly, the Harmonization of Eczema Outcome Measurements initiative, spearheaded by an international consensus group, recommends a core set of measurement tools that encompass both the objective clinician-driven parameters and patient-oriented instruments incorporated into clinical practice (Table 3) [35]. Therefore, it is imperative that patients be fully informed of the advantages and disadvantages of any systemic therapy prior to selecting a therapeutic option. As biologics are monoclonal antibodies designed to target specific cytokines or immunomodulatory proteins,they tend to be more selective than small-molecule inhibitors; moreover, they have a longer history with more enriched long-term and real-world data. On the other hand, their onset of action tends to be slower. As small-molecule inhibitors target signaling pathways, they are less selective than biologics. However, their anti-inflammatory action is broader in spectrum, which may aid the treatment of AD, which has a multifaceted immunopathogenesis. The oral administration route is also useful for treating patients who are averse to injections, and the faster onset of action facilitates its usage for flare-ups. On the other hand, fewer long-term and real-world data are available for small-molecule inhibitors due to their shorter history of development; thus, caution must be exercised with due vigilance about safety concerns.
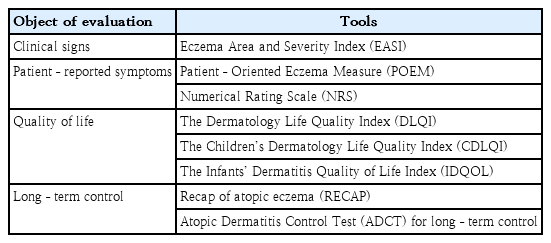
Harmonization of eczema outcome measurements initiative recommendations about atopic dermatitis control measurement tools by objective
Aside from choosing the optimal therapy tailored to each patient's individual phenotype, it is imperative that the basic principles, including providing patient education, such as how to monitor and manage a skin barrier and avoid exacerbating factors, ensuring the appropriate use of local therapy, and implementing a rigorous monitoring regiment suitable for pediatric patients, not be neglected (Table 3).
Conclusion
Pediatric AD is a systemic disease that marks the beginning of the allergic march, and its severity is correlated with the risk of further evolution of allergic comorbidities. Considering the significant impact of AD on the physical and mental health of the patient and their family as well as the burden of healthcare costs that befalls society, the importance of early and effective treatment of AD cannot be overstated.
To fully leverage the recent technological and therapeutic advancements, systemic changes must also be enforced. The appropriation and implementation of reasonable service fees allocated to treatment and education as well as nationwide support of identifying novel therapies are paramount. Further research on the ideal dosing, usage, and monitoring schedule will continue to optimize treatment outcomes.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.