Comprehensive evaluation of the child with global developmental delays or intellectual disability
Article information

Abstract
Global developmental delay (GDD) and intellectual disability (ID) are relatively common neurodevelopmental disorders that significantly impact affected children, their families, and society. The etiology of GDD/ID is notably diverse, encompassing both genetic and acquired factors. Although the precise cause of most GDD/ID cases remains unclear, an estimated half of all cases can be attributed to genetic factors. Thus, a detailed medical history and comprehensive physical examination remain pivotal for guiding diagnostic investigations into the underlying causes of GDD/ID. Advancements in genetic testing have supplanted traditional methods such as karyotyping and fluorescence in situ hybridization with chromosomal micro arrays, which are now the primary genetic tests for children with idiopathic GDD/ID. Moreover, the evaluation of Fragile X and Rett syndrome should be an integral component of initial diagnostic assessments. In recent years, whole-exome sequencing and whole-genome sequ-encing have emerged as important diagnostic tools for evaluating children with GDD/ID and have substantially enhanced the diagnostic yield rates. Gene therapy has emerged as a promising avenue and is poised to become a cornerstone in addressing various genetic developmental and epilepsy disorders. Early intervention facilitated by a proficient multidisciplinary team can markedly enhance the prognosis and outcomes of GDD/ID, particularly when parents or caregivers are actively engaged in the interventional process. This review discusses risk factors and common underlying causes, explores recent evidence and recommendations for genetic evaluation, and offers management strategies for children with GDD/ID.
Key message
· A detailed history and comprehensive physical examination remain the cornerstones for establishing a diagnosis of global developmental delay/intellectual disability (GDD/ID).
· Comprehensive surveillance and screening programs play a significant role in the early detection of GDD.
· Whole-exome sequencing is highly recommended as first- or second-line testing for individuals with idiopathic GDD/ID.
· Early intervention by a well-versed multidisciplinary team can significantly improve the outcomes and prognosis of GDD/ID.
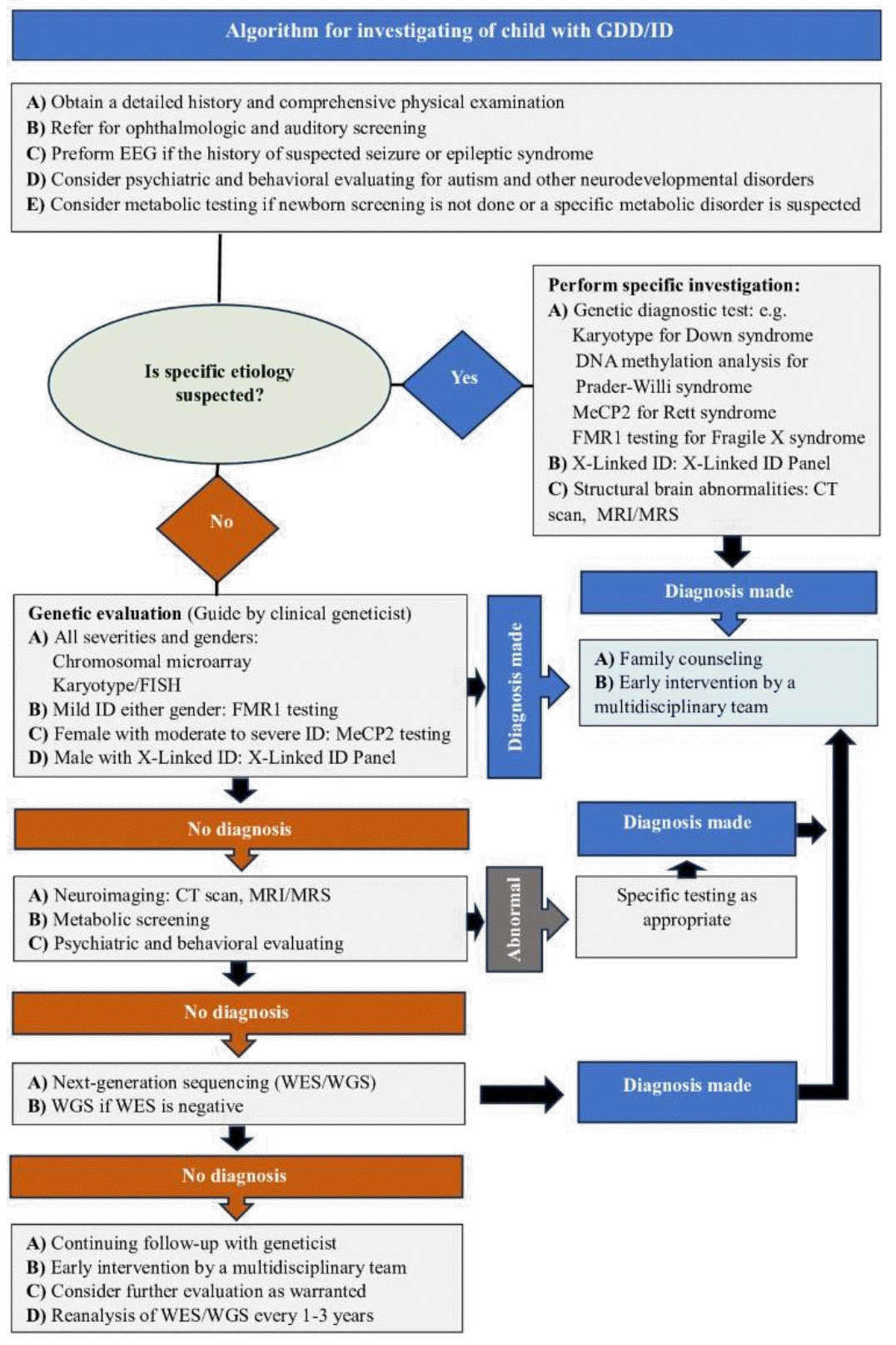
Graphical abstract. Algorithm for investigating of child with GDD/ID. GDD, global developmental delay; ID, intellectual disability; EEG, electroencephalography; MeCP2, methyl-CpG binding protein 2; FISH, fluorescence in situ hybridization; CT, computed tomography; MRI, magnetic resonance imaging; MRS, magnetic resonance spectroscopy; WES, whole-exome sequencing; WGS, whole-genome sequencing.
Introduction
Developmental delay (DD) is among the most prevalent chronic medical conditions encountered in general pediatric, neurological, and genetic clinics. DD refers to one's inability to achieve developmental milestones within an anticipated timeframe across one or more domains including motor skills, language, cognition, social interaction, emotional development, and adaptive functioning [1]. DD can manifest as isolated delays within a specific domain or as global DD (GDD), which is characterized by delays equal to 2standard deviations below the mean in 2or more developmental domains among children under 5 years of age [1]. Furthermore, DD can exhibit transient features, particularly in cases of chronic illness, or persist, potentially culminating in developmental disabilities [2].
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) defines intellectual disability (ID) as one of several neurodevelopmental disorders (NDDs) that begin in childhood. ID is characterized by deficits in intellectual functioning (problem-solving, reasoning, academic learning, planning, abstract thinking, judgment, and learning from experience) and difficulties in adaptive functioning (competence in social, conceptual, and practical skills) [1]. NDDs are a group of cognitive and behavioral disorders that become apparent in early childhood and are characterized by evident impairments or limitations in personal, academic, social, or occupational functioning. This category encompasses GDD and ID as well as other conditions such as cerebral palsy (CP), autism spectrum disorder (ASD), attention-deficit hyperactivity disorder (ADHD), learning disabilities, developmental language impairments, and developmental coordination disorder [1]. The overall prevalence of DD and ID varies and is influenced by numerous factors including socioeconomic status, environmental conditions, and assessment methodologies.
Community-based studies conducted in various countries reported that the prevalence of DD in children is 10%–15%, whereas GDD affects approximately 1%–3% of children. ID affects an estimated 1%–3% of school-aged children, with approximately 85% of cases classified as mild, and many individuals in this category even achieve academic success [2,3]. Several studies have suggested that a family's medical history could play a role in GDD, ID, and NDDs, especially when other family members are similarly affected. These conditions may encompass genetic or chromosomal abnormalities such as Down syndrome or Fragile X syndrome as well as NDDs such as ADHD and ASD. Although the precise prevalence of GDD/ID within families remains uncertain, various studies reported rates of 12.5%–22.5% [2,4,5].
DD does not always signify a developmental disability. However, it can serve as an early indicator of learning disabilities, and many children with disabilities encounter various DDs. DD severity is typically categorized as mild, moderate, or severe based on functional age as assessed by a developmental pediatrician using age-appropriate standardized measures of function and development compared with chronological age (i.e., mild, 67%–100%; moderate, 33%–66%; severe, <33%) [2,6]. Intellectual functioning is commonly evaluated using intelligence quotient (IQ), with an average of 100. Children with ID typically exhibit IQ scores <70 (2 standard deviations below the average) [7]. However, not all children with GDD have concurrent deficits in intellectual functioning. Research indicates that approximately 20% of preschoolers with GDD demonstrate average intellectual functioning [8]. It is important to recognize that not all children with GDD develop ID later in life. Developmental disorders such as CP are frequently associated with notable delays in motor skills, speech, and cognition. Nevertheless, some individuals may demonstrate average intelligence [9].
GDD/ID has various causes that are typically categorized into acquired and genetic. Although the exact cause is often unknown, genetic factors contribute to approximately half of all cases [2,10]. Genetic testing has become an essential component of the diagnostic evaluation. The early identification of warning signs and underlying causes, along with the implementation of appropriate management strategies, is crucial for predicting the patient's clinical course, enhancing their intellectual functioning and developmental skills, and providing accurate prognostic counseling. This study aimed to review the risk factors, etiologies, diagnostic approaches (including available genetic tests and their results), and management strategies for children with GDD/ID.
Risk factors and etiologies
Multiple risk factors contribute to GDD/ID, including premature birth, low birth weight, perinatal complications, chronic illness, chronic malnutrition, low socioeconomic status, and a lack of appropriate care [11]. GDD/ID has a heterogeneous etiology that encompasses both acquired and genetic causes. Although the etiology of most GDD/ID cases is unknown, a genetic origin is responsible for up to 50% of cases. Down syndrome is the most common chromosomal abnormality associated with GDD/ID, whereas Fragile X syndrome is the most prevalent genetic disorder. Other possible causes include Rett syndrome,Angelman syndrome, Williams syndrome, Prader-Willi syndrome, and subtle translocations or deletions in genetic material [12,13].
Metabolic disorders represent a relatively uncommon etiology of GDD/ID, comprising only approximately 1%–5% of cases. Furthermore, they typically involve multiple organ systems and manifest as acute decompensation or regression of developmental milestones rather than mere delays. However, it is imperative to consider metabolic disorders such as congenital hypothyroidism, phenylketonuria, and methylmalonic acidemia as potential underlying causes of GDD/ID [14,15]. In addition to central nervous system malformations, brain injury leading to neurodevelopmental disabilities can stem from pre- and postnatal complications such as asphyxia, congenital and acquired infections, kernicterus, and head trauma [16,17]. Uncontrolled maternal medical conditions and environmental exposure during pregnancy can precipitate GDD/ID. Such exposures may include the maternal consumption of alcohol, opioids, and teratogenic medications [18,19]. Premature infants or those with nutritional deficiencies, chronic illness, familial stress, immaturity, or prolonged hospitalization may experience transient DDs [2]. Table 1 lists the most common causes of GDD/ID [2,5,12-19].

Etiologies of global developmental delay and intellectual disability
Diagnostic approach
Developed countries have well-established healthcare systems that employ comprehensive surveillance and screening programs to facilitate the early recognition and intervention and improve the outcomes of children with GDD/ID. Clinicians must be familiar with the normal developmental milestone ranges and the evolving maturation process to promptly recognize delays [20]. Moreover, it is important to adjust the chronological ages of premature infants to appropriate milestones by considering their corrected gestational ages. The diagnosis of GDD/ID requires an intellectual assessment, adaptive evaluation, audiometry, vision testing, and psychiatric and behavioral assessments [2].
Several intelligence tests have been developed to assess mental abilities, including abstract thinking, learning, and problem-solving in novel situations. Commonly utilized tests include the Wechsler scale, Stanford-Binet Intelligence Scale, and Differential Ability Scales. The IQ score is frequently used to compare an individual's intellectual capacity to the average score of a comparable group. Based on IQ score, individuals can be categorized into different ID levels: Mild ID (IQ 50–70) is the most prevalent, accounting for 85% of cases, followed by moderate ID (IQ 35–50), which is observed in 10% of cases, and severe ID (IQ 20–35), which is present in 4% of cases. Profound ID (IQ <20) is the least common, occurring in only 1% of cases [7,21]. The diagnostic process for GDD/ID typically commences with a thorough clinical history, comprehensive physical examination, and behavioral assessment, which requires further investigation.
1. History and physical examination
A thorough clinical history and physical examination are essential to diagnostic accuracy, as they can reveal the underlying cause of the disorder in up to 38.6% of cases [22]. To ensure diagnostic accuracy, it is crucial to gather detailed information about the patient's gestation, prenatal and perinatal events, delivery mode, neonatal care, developmental milestones, medical history, psychosocial background, maternal health, consanguinity, and three-generation family history. A meticulous neurodevelopmental assessment may also provide additional insight into the common causes of GDD/ID, including the evaluation of developmental milestones across all domains, growth parameters, and neurological findings.
Growth parameters (especially head circumference) should be plotted on appropriate growth charts and compared with previous measurements and with those of other family members [2,7,12,17]. The presence of dysmorphic features indicates specific genetic syndromes. However, in some children with dysmorphic features and/or malformations with or without GDD/ID, a detailed history and comprehensive examination may not immediately suggest a diagnosis. Therefore, the test should be conducted by a trained clinical geneticist, particularly in cases with subtle findings that are often important for the differential diagnosis. An essential component of the dysmorphological examination is observation of the patient from head to toe as well as a comparison with established normative values and photographs of the parents and siblings. Many children with dysmorphic features have DD, malformations, or both. A large study of individuals with NDDs identified dysmorphic features in approximately 47% of cases [23].
Various syndromes with distinct dysmorphic features can also result in GDD/ID; thus, clinicians must be knowledgeable and acquainted with these conditions. Fragile X syndrome, among the most prevalent of these syndromes, is characterized by identifiable clinical features that are easily noted in affected males with the full mutation. These features encompass dysmorphic facial characteristics (large ears, elongated faces, pronounced jaws, macrocephaly, hyperextendable joints, soft skin, and hypotonia) along with macroorchidism. However, these features may be subtle, particularly in females or prepubertal males.
Furthermore, Rett syndrome, an X-linked dominant disorder that affects primarily girls, demonstrates variable clinical manifestations marked by a progressive cognitive decline, loss of purposeful hand function accompanied by stereotypical hand movements (such as clapping, wringing, and tapping), speech and language impairments, acquired microcephaly, and progressive lower-limb spasticity. Breathing abnormalities such as breath-holding, hyperventilation, and air swallowing are commonly observed. Additionally, gait apraxia, kyphoscoliosis, sleep disturbances, episodes of laughter and cream, and a reduced response to pain are other variable characteristics of this disorder [2,12,23,24].
Anomalies affecting the musculoskeletal system, eyes, heart, genitourinary tract, and connective tissues may also be discerned during routine systemic physical examinations, potentially those indicating a specific diagnosis or guiding laboratory investigations. Behavioral assessment provides the basis for describing any atypical pattern of a child’s behavior, such as aggression, repetitive and self-injurious behavior, decreased social engagement, and impulsivity. Table 2 outlines the essential points of history taking and physical examination.

Key points in history taking and physical examination
2. Biochemical and metabolic investigations
In addition to paying careful attention to resource utilization and risk-benefit analyses, laboratory evaluations should be carefully selected. Given that many cases of GDD/ID are idiopathic and significant or pathognomonic findings in laboratory tests are uncommon, it is imperative that tests be conducted judiciously to prevent unwarranted investigations [7,22]. In developed countries, all newborns undergo screening programs at birth that encompass critical cardiac diseases, hearing loss, and a spectrum of inheritable disorders related to metabolic, hematologic, endocrine, and other conditions. Consequently, it is inadvisable to conduct these screening tests in children with GDD/ID unless a specific underlying cause is indicated by their medical history or physical examination findings [25].
An initial assessment may include routine complete blood count, electrolytes, basic chemistry and lipid panels, iron, creatinine kinase, thyroid function, and liver function tests. A thorough investigation of inborn errors of metabolism may involve plasma amino acids, ammonia, lactic acid, serum lactate/pyruvate, urine organic acids, capillary blood gases, and lysosomal enzymes. Additional tests, if warranted, could encompass uric acid levels, very long-chain fatty acids, transfer, copper, ceruloplasmin, and acylcarnitine levels. A routine urinalysis is performed to detect oligosaccharides, glycosaminoglycans, sialic acids, urine purines, urea, and pyrimidines. A cerebrospinal fluid analysis is not recommended for the routine evaluation for GDD/ ID. However, specific indications including glucose, lactic acid, glycine, and pyruvate may be considered.
Screening for congenital infections (e.g., Toxoplasma, rubella, cytomegalovirus, hepatitis B virus, human immunodeficiency virus,herpes virus) may also be warranted in cases with symptoms of microcephaly, neurological anomalies, and hearing or vision loss. Screening for lead toxicity should focus on children with identifiable risk factors for environmental exposure. Furthermore, more comprehensive testing for specific diseases is typically justified once the results become available [2,7,10,12,17].
3. Neuroimaging and electroencephalography
Although routine neuroimaging studies are not recommended for assessing children with unexplained GDD/ID, their use may lead to the recognition of a specific cause, particularly in children who present with dysmorphic features or focal neurological findings [26]. Brain magnetic resonance imaging (MRI) has a varied diagnostic yield of 47%–72% depending on the use of newer techniques and additional clinical findings. MRI is more sensitive than computed tomography at detecting clinically relevant structural abnormalities and anomalies related to myelination and neuronal migration, resulting in a higher diagnostic yield [26,27]. Electroencephalography may play an important role in the assessment of children with GDD/ID, as it helps clinicians classify and evaluate seizures and identify specific epileptic syndromes, such as acquired epileptiform aphasia syndrome and electrical status epilepticus, during slow-wave sleep. However, its use is not routinely recommended as part of the initial evaluation unless there are historical features suggestive of epilepsy, a specific epileptic syndrome, or speech regression [12].
4. Genetic testing
Genetic disorders and congenital anomalies contribute significantly to childhood mortality, morbidity, and lifelong disabilities. Over the past three decades, advances in genetics have revolutionized human health by introducing novel testing approaches to facilitate diagnosis, treatment, therapy, improved outcomes, preventive screenings, and population-based risk assessments. Notably, up to 80% of rare disease cases have identifiable genetic origins, underscoring the pivotal role of genetic tests and their impact on patients, families, and the healthcare system [28,29]. Table 3 summarizes the recent studies that highlighted the genetic significance of assessing individuals with GDD/ID and NDDs [30-45].
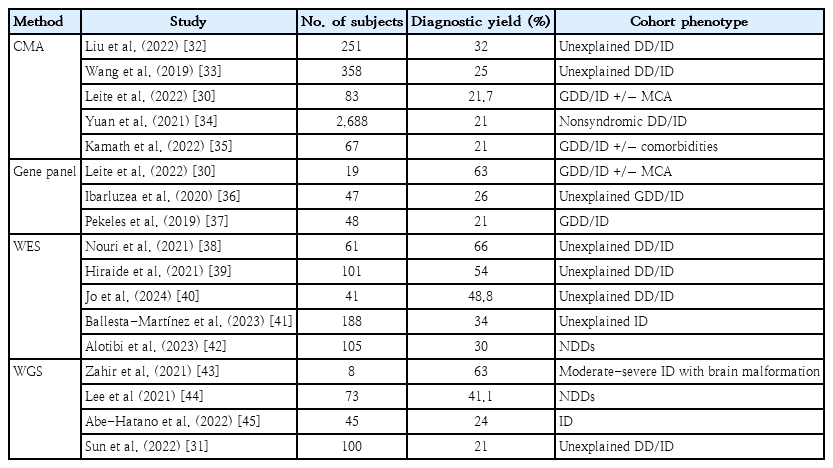
Recent studies highlighting genetic significance of assessing individuals with GDD/ID
1) Chromosome microarray analysis
Genetic etiologies, including chromosomal abnormalities (e.g.,microdeletions,microduplications, and trisomies),have been proposed to account for approximately half of GDD/ID cases [2,10]. Copy number variations, genetic deletions and duplications, and encompassing insertions are important contributors to human genetic diversity and sources of associated genetic mutations. The enhanced sensitivity of microarray-based genomic copy number analysis enables the detection of submicroscopic deletions and duplications, resulting in a significantly higher diagnostic yield than that of karyotyping (15% vs. 7.4%, respectively). Thus, it has become a standard diagnostic genetic test for patients with unexplained GDD, ID, ASD, epilepsy, or multiple congenital anomalies. Furthermore, it is the most effective diagnostic tool for evaluating GDD/ID after a thorough history and comprehensive physical examination [46-48].
In one study, a chromosome microarray analysis (CMA) was performed in patients with normal karyotype results, resulting in a diagnostic yield of 21.7% (18 of 83) [30]. Two CMA platforms were used to analyze chromosome array comparative genomic hybridization and single-nucleotide variant arrays. These platforms are utilized to visualize and detect chromosomal copy number variants, including genomic gains (microduplications) and losses (microdeletions), with a higher resolution for genomic imbalances than conventional G-banded karyotypes. However, CMA has limited ability to detect low-level mosaicism and cannot detect balanced chromosomal rearrangements [49,50].
Patients with both syndromic and nonsyndromic GDD/ID can exhibit a wide range of phenotypic features caused by various microdeletions and duplications. Some examples of such syndromes include Prader-Willi and Angelman syndromes (deletion of 15q11-q13), Smith-Magenis syndrome (deletion of 17p12), Williams-Beuren syndrome (deletion of 7q11.23), and deletions in the CHRNA7 and SH2B1 genes, among others [2,7,10,12]. Interpreting variants of uncertain significance identified by CMA can be challenging, particularly when identifying incidental findings, such as the deletion of cancer predisposition genes such as FLCN or BMPR1A, which can have significant ethical implications, especially when investigating GDD/ID. Therefore, it is recommended that the interpretation of abnormal results and variants of unknown significance in the CMA test and subsequent counseling of families be performed by a medical geneticist and certified genetic counselor in collaboration with the reference laboratory and platform used. This will ensure that the interpretation is accurate and appropriate counseling is provided to families [51].
2) Chromosomal karyotyping
Karyotyping is considered the first standard diagnostic test for children with GDD/ID for over 35 years. However, it has been replaced by CMA, which can perform a similar function at much higher resolution for genomic imbalances, thus substantially increasing sensitivity. Although karyotyping has the limitations of low resolution and a detection rate of approximately 7.4% for evaluating patients with unexplained GDD/ID, it has been recommended instead of CMA in cases of clinically suspected aneuploidy (e.g., Down syndrome, Turner syndrome), multiple spontaneous abortions, refractory epilepsy, and a family history of chromosomal rearrangements [30,48].
3) Fragile X and rett syndrome testing
Fragile X syndrome is an X-linked trinucleotide expansion disorder caused by the unstable expansion of a CGG triplet repeat sequence in the FMR1 gene on Xq27.3. Fragile X syndrome is the most common genetic abnormality associated with ID and the most common monogenic cause of ASD [52]. The prevalence of Fragile X syndrome varies among individuals with GDD/ID. An estimated 2.2%–2.5% of affected individuals with GDD/ID caused by Fragile X syndrome are boys, whereas 1.3%–1.6% are girls [50]. Although Fragile X syndrome may occur in both males and females, its distinctive clinical features are most easily recognized in males with the full mutation. However, this effect may be subtle or apparent only upon puberty [52].
The diagnostic yield of Fragile X testing among patients with NDDs varies widely depending on factors such as the characteristics of the population being tested, study design, and sex. This yield is estimated to be 1.2%, with significantly higher rates observed when testing is restricted to males exhibiting NDD-consistent physical and behavioral phenotypes, particularly in the presence of a positive family history [53]. Fragile X should be strongly considered and recommended as part of first-line investigations in both boys and girls with GDD/ID and/or ASD, especially in the presence of a positive family history, consistent physical and behavioral phenotype, and the absence of major structural abnormalities. Use of the X-linked ID panel may be appropriate for patients with a family history of X-linked inheritance and normal microarray, chromosomal, or Fragile X results [2,7,12,53].
Rett syndrome is a rare and severe NDD that almost exclusively affects girls and is caused by mutations in the X-linked gene encoding methyl-CpG binding protein 2 (MECP2) on the long arm of the X chromosome (Xq28) [54]. A recent meta-analysis estimated the global prevalence of Rett syndrome as 7.1 cases per 100,000 female individuals [55]. Rett syndrome has variable clinical expressions but is characterized by progressive cognitive impairment, speech and language disabilities, decelerated head growth (acquired microcephaly), and loss of purposeful hand use, which is often replaced with stereotypic hand movements [54]. In contrast, mutations of MECP2 in boys may result in severe neonatal encephalopathy [56]. Notably, some patients with Rett syndrome do not harbor mutations in the MECP2 gene. Interestingly, others with mutations in the MECP2 gene do not have the classical clinical features of Rett syndrome [57]. However, when Rett syndrome is clinically suspected, a molecular analysis should be performed.
4) Next-generation sequencing
Next-generation sequencing (NGS) effectively identifies novel gene mutations and discrete genomic variations, hence increasing the genetic diagnostic yield of GDD/ID [58]. Whole-exome sequencing (WES) can be used concurrently to interpret the DNA sequences of thousands of protein-encoding genes using NGS techniques. WES is proven helpful in patients with multiple genetic conditions, atypical presentations of a genetic disorder, and early presentations of a disease for which classic findings are lacking. However, WES has important limitations, one of which is incomplete exome coverage, which does not allow for the examination of all exons potentially affecting the test yield. Moreover, WES does not reliably detect mosaic variants, repetitive sequences, exon-level deletions, epigenetic variants, or balanced rearrangements [59]. In a recent meta-analysis focusing on isolated NDDs, the estimated diagnostic yield of WES was 31%, greater than that of CMA (15%). Higher yields were observed in patients with abnormal head size, younger age at presentation, and developmental epileptic encephalopathy [49]. According to the 2021 American College of Medical Genetics and Genomics guidelines, WES is highly recommended as a first- or second-line test for children with GDD/ID [49,60].
Whole-genome sequencing (WGS) is an NGS approach that enables the in-depth analysis of entire genomes, including exons, noncoding DNA, and structural variants. WGS is unavailable for clinical use in most medical centers and restricted to clinical research. According to a recent review, the diagnostic yield of WGS in cases of GDD/ID is 21%–64% [31]. In a clinically heterogeneous cohort, WGS as the main clinical test provided a greater diagnostic success rate than conventional genetic testing [61]. Overall, compared to WES, extrapolation of the additional information by WGS was estimated to improve the diagnostic rate of WES by 9%–15% [62]. Certain genetic disorders are not evident at birth or during the first few years of life. Thus, continued follow-up by geneticists and an NGS reanalysis every 1–3 years are recommended to enhance the diagnostic yield by incorporating new disease gene discoveries, additional phenotypic information becoming available over time, variant reclassification, and improved bioinformatics pipelines. The diagnostic benefits of a periodic WES and WEG data reanalysis have shown improvements in diagnostic yield of 12.6% and 10.9%, respectively, in individuals with NDDs [63,64].
5. Hearing and visual assessments
Since children with GDD/ID are more susceptible to visual and hearing abnormalities, it is crucial to provide them with a comprehensive ophthalmological assessment, including a dilated funduscopic examination and vision screening. Furthermore, behavioral observation audiometry and brainstem auditory-evoked responses are recommended, particularly for children exhibiting speech and language delays or autistic characteristics [65-67]. Table 4 outlines the essential investigations for evaluating individuals with GDD/ID.

Comprehensive workup of children with GDD/ID
Management and outcome
The primary principle in managing individuals with GDD/ID is early intervention, which not only enhances their developmental functioning and potential but improves their quality of life and social participation [68]. Providing multidisciplinary services and implementing structured school and home exercise programs can positively impact the psychosocial well-being of children and their caregivers [67]. Numerous studies have shown that involving parents or caregivers in early interventions leads to functional improvements in individuals with NDDs [69,70]. Individuals with GDD/ID may have various physical, behavioral, and mental-health comorbidities, including epilepsy, vision and hearing impairments, ADHD, aggressive or self-injurious behavior, sleep disturbances, obstructive sleep apnea, recurrent chest infections, malnutrition, constipation, gastroesophageal reflux disease, movement disorders, and orthopedic deformities [71,72]. Individuals with GDD/ID must be evaluated by a multidisciplinary team that is familiar with these disorders and their associated complications. Such teams may comprise pediatricians, neurologists, child and adolescent psychiatrists, developmental and behavioral pediatricians, audiologists, ophthalmologists, speech therapists, physical therapists, occupational therapists, orthopedic surgeons, and other relevant pediatric subspecialists as required [73,74].
Although no specific medical therapy is available for most of GDD/ID cases, certain instances can be reversible or treatable if identified promptly and managed effectively. These include acquired and inherited metabolic disorders such as homocystinuria, methylmalonic aciduria, biotinidase deficiency, hypothyroidism, and nutritional deficiencies, among other conditions [75]. Furthermore, managing associated comorbidities such as epilepsy, spasticity, aggressive or self-injurious behavior, and sleep disorders may necessitate appropriate pharmacological interventions [76].
In recent years, gene therapy has emerged as a promising solution for addressing a variety of genetic, developmental, and epileptic disorders that can significantly affect an individual's cognitive and physical development. This innovative approach targets the underlying genetic causes of these conditions and often utilizes viral vectors to deliver corrected genes or gene editing tools directly into patient cells. For example, in disorders such as Rett syndrome and Fragile X syndrome, gene therapy has shown promising ability to improve motor and behavioral symptoms in animal models. However, challenges remain in determining optimal expression levels and mitigating the associated risks.
Alternative strategies, such as clustered regularly interspaced short palindromic repeats-mediated gene editing and antisense oligonucleotides, are being explored to address these challenges. Additionally, gene therapy approaches for disorders such as Angelman syndrome and Dravet syndrome aim to enhance specific gene activity to counterbalance genetic mutations. Clinical trials have demonstrated improved cognitive functions, motor skills, and overall quality of life in patients with genetic developmental disorders or epilepsy. Despite ongoing challenges related to safety, efficacy, and ethical considerations, the clinical application of gene therapy represents a significant advancement in personalized and transformative treatments for these complex conditions [77]. Finally, the early involvement of a geneticist can help with diagnostic confirmation, workup guidance, and genetic counseling. In some cases, social work and family support services can offer counseling, monetary, and educational support as well as connections to other support services to help families manage their child's condition.
Conclusion
Comprehensive surveillance and screening programs play a crucial role in the early detection of GDD/ID in children by facilitating interventions that can significantly enhance outcomes. Guidelines for evaluating children with GDD/ID are generally similar, commencing with a thorough history taking, followed by a comprehensive examination, which serves as the foundation for planning diagnostic investigations aimed at identifying the etiology. Genetic testing is a crucial and highly effective tool for diagnosing GDD/ID. However, it is essential to recognize that each genetic diagnostic method has its advantages and limitations, which necessitates careful consideration based on specific clinical scenarios.
It is imperative to acknowledge that there is no one-size-fits-all approach and that the decision-making process should prioritize clinical suspicion and the objectives of genetic testing. CMA is an established diagnostic test for children with unexplained GDD/ID. Nevertheless, WES and WGS have demonstrated greater effectiveness at identifying GDD/ID etiology, particularly in cases of complex clinical phenotypes or uncertainty regarding the underlying genetic cause. Consequently, they are now recommended as first- or second-tier approaches to examining unexplained GDD/ID. The clinical application of gene therapy represents a paradigm shift in the treatment of various genetic disorders including GDD/ID. By targeting the underlying genetic causes of these conditions, gene therapy offers the potential for improved outcomes and enhanced quality of life in affected individuals. As research progresses and technology evolves, gene therapy has become a cornerstone in the management of GDD/ID. Although most GDD/ID cases are incurable, early supportive management and therapies such as physical, speech, and occupational therapies are effective and should be encouraged to maximize the abilities of an affected child.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.