Applications of genomic research in pediatric endocrine diseases
Article information

Abstract
Recent advances in molecular genetics have advanced our understanding of the molecular mechanisms involved in pediatric endocrine disorders and now play a major role in mainstream medical practice. The spectrum of endocrine genetic disorders has 2 extremes: Mendelian and polygenic. Mendelian or monogenic diseases are caused by rare variants of a single gene, each of which exerts a strong effect on disease risk. Polygenic diseases or common traits are caused by the combined effects of multiple genetic variants in conjunction with environmental and lifestyle factors. Testing for a single gene is preferable if the disease is phenotypically and/or geneically homogeneous. Next-generation sequencing (NGS) can be applied to phenotypically and genetically heterogeneous conditions. Genome-wide association studies (GWASs) have examined genetic variants across the entire genome in a large number of individuals who have been matched for population ancestry and assessed for a disease or trait of interest. Common endocrine diseases or traits, such as type 2 diabetes mellitus, obesity, height, and pubertal timing, result from the combined effects of multiple variants in various genes that are frequently found in the general population, each of which contributes a small individual effect. Isolated founder mutations can result from a true founder effect or an extreme reduction in population size. Studies of founder mutations offer powerful advantages for efficiently localizing the genes that underlie Mendelian disorders. The Korean population has settled in the Korean peninsula for thousands of years, and several recurrent mutations have been identified as founder mutations. The application of molecular technology has increased our understanding of endocrine diseases, which have impacted on the practice of pediatric endocrinology related to diagnosis and genetic counseling. This review focuses on the application of genomic research to pediatric endocrine diseases using GWASs and NGS technology for diagnosis and treatment.
Key message
· Recent advances in molecular genetics have improved our understanding of pediatric endocrine disorders and are now used in mainstream medical practice.
· Genome-wide association studies can increase our understanding of the biological mechanisms of disease and inform new therapeutic options.
· The identification of founder mutations leads to the efficient localization of the genes underlying Mendelian disorders.
· Next-generation sequencing technologies benefit clinical practice and research of pediatric endocrinology.
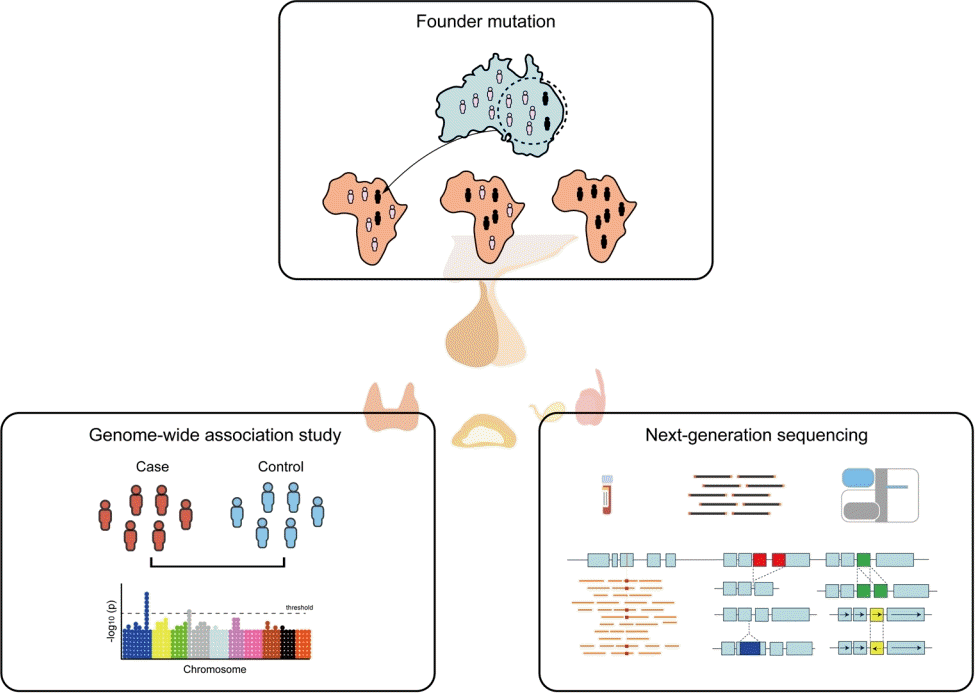
Graphical abstract
Introduction
Pediatric endocrine diseases have a substantial genetic component. Identifying the genetic etiologies of these diseases helps researchers identify their molecular pathophysiology and clinicians provide customized treatment and disease prevention. Many pediatric endocrine disorders are caused by defects in genes involved in hormone synthesis, binding proteins, transcriptional activity, channels, membrane receptors, and signal transductions [1]. Genetic syndromes in endocrine diseases are based on a unique genetic pathophysiology.
There are 2 extremes in the spectrum of endocrine genetic disorders: Mendelian and polygenic. Mendelian or monogenic diseases, which represent the extremes of possible genetic architecture, are caused by rare sequence variants in a single gene, each of which has a large effect on disease risk (Fig. 1) [1]. Monogenic disorders, such as congenital adrenal hyperplasia, are inherited conditions arising from mutations in a single gene that disrupt a physiological pathway [2,3]. Genetic variants that cause Mendelian diseases occur in a few genes, can be highly penetrant, and are nearly unaffected by the environment. The wide spectrum of phenotypes that characterize many Mendelian endocrine diseases is often reflected in genetic heterogeneity. Some Mendelian disorders demonstrate the same phenotype owing to multiple mutations in the same gene or locus, known as allelic heterogeneity. Others present as the same disease in different individuals and result from mutations in multiple different genes, known as locus heterogeneity; one example of this is Kallmann and Noonan syndromes [2].
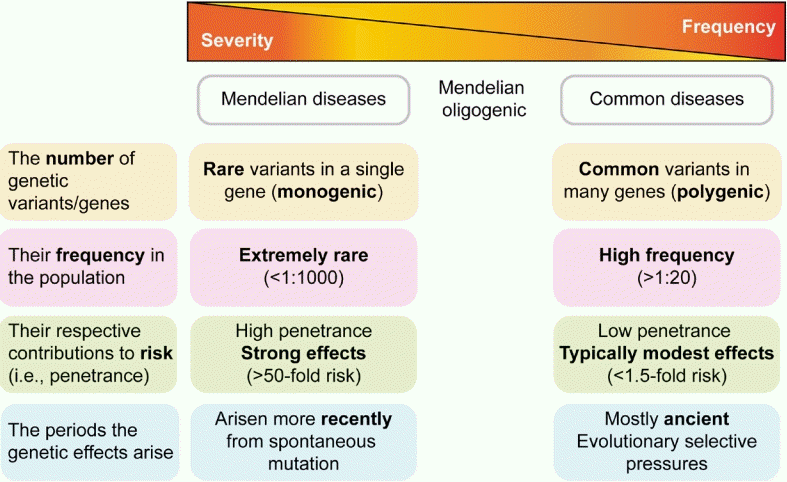
Spectrum of genetic impacts/architectures in Mendelian and polygenic disorders. Mendelian diseases are caused by rare variants in a single gene, each with a strong effect on disease risk. Common diseases and traits are caused by the combined effects of multiple variants observed frequently in the population, each with a modest effect. Common variants are mostly ancient and typically have relatively modest clinical effects, whereas rare variants tend to have arisen more recently and can exert larger clinical effects.
At the other extreme of the spectrum are common or polygenic diseases and traits such as type 2 diabetes mellitus (DM), obesity, or human height, which are caused by the combined effects of multiple common variants and frequently observed in the population, with small to modest effects in conjunction with environmental and lifestyle factors (Fig. 1) [3].
The identification of disease genes in rare, monogenic, or syndromic disorders has mostly been driven by linkage analysis within multiple pedigrees [3]. This approach has been successful for identifying high-penetrant variants responsible for Mendelian disease [3]. In contrast, variants of common polygenic disorders have been identified in genome-wide association studies (GWASs) in the general population [4].
Diagnosing endocrine diseases requires comprehensive tools, including medical information, biochemical tests, dynamic hormone tests, and imaging studies. Genetic testing is currently used as a confirmatory diagnostic tool for many pediatric endocrine diseases, particularly if the biochemical findings are vague. Over the last 2 decades, advancements in molecular genetic technologies have led to profound development in the diagnostics and research of pediatric endocrine diseases. Molecular techniques for genetic diseases have developed from traditional methods to modern genetic testing [2]. Traditional genetic testing includes routine karyotype analysis or tests for single genes or chromosomal regions, such as Sanger sequencing or fluorescent in situ hybridization. Modern genetic testing analyzes the entire genome using technologies such as chromosomal microarray and next-generation sequencing (NGS) [2].
Knowledge of the contribution of genetic and epigenetic alterations to endocrine disorders has massively expanded and furthered our understanding of their molecular pathophysiology, the provision of adequate genetic counseling and prenatal diagnosis, and the development of new therapeutic strategies based on the understanding of the molecular mechanisms of such diseases. This review focuses on the application of genomic technologies to pediatric endocrine diseases, such as GWASs and founder mutations. In addition, we highlight the benefits of moving from traditional genetic testing to NGS technologies to diagnose and establish treatment strategies for pediatric endocrine diseases.
GWASs for pediatric endocrine diseases
GWASs aim to determine statistically associated genetic variants across the entire genome in a large number of individuals matched by population ancestry and evaluate the disease or trait of interest [4]. GWASs can provide genetic information for use as screening tools to identify individuals at risk of certain diseases or conditions. The most commonly studied genetic variants in GWASs are single-nucleotide polymorphisms (SNPs) [4]. The primary goals of these studies were to better understand the disease biology and provide prevention or treatment strategies. Although a genetic variant at a certain locus and trait is not directly informative, GWASs have been successfully implemented to predict the relative roles of genes and the environment in disease risk [5].
GWASs are based on the common variant, common disease hypothesis, which holds that the genetic basis for common diseases and traits is driven by common genetic variants [4]. The statistical power to detect associations between genetic variants and a trait requires very large sample sizes because of the small effect sizes of variants in GWASs. Using millions of association tests, a strict statistical threshold must be set to establish significance at a genome-wide level, which corresponds to a P value of <5×10-8 [5]. However, the vast majority of SNPs are novel and located in noncoding regions, making it difficult to interpret the functional consequences of the variant on the phenotype [5]. In addition, well-validated cell and animal models to test its functional impact are lacking. Therefore, a diverse set of approaches has been developed to infer the functional impacts of variants identified by GWASs. For coding variants, multiple annotation tools, such as ANNOVAR or variant effect predictor [6], can infer their potential impact on genes; however, only 2%–3% of the loci are present in this region. Expression quantitative trait loci analysis, which identifies loci associated with RNA expression, can be used to identify the functional impact on the regulatory region [7]. Various projects for mapping the effects of regulatory variations, such as ENCODE, Roadmap Epigenomics, and the Genotype-Tissue Expression project, provide an essential dissection of the characterization and interpretation of noncoding variants [7,8].
Common endocrine diseases and traits, such as type 2 DM, obesity, height, and puberty, result from the combined and simultaneous effects of multiple variants in various genes, commonly found in the general population, with each contributing a small effect. Polygenic risk scores (PRSs) can be calculated using the small effects of a number of genetic variants discovered in GWASs. These scores can predict the risk of complex diseases, such as coronary artery disease, atrial fibrillation, type 2 DM, inflammatory bowel disease, and breast cancer [9]. PRSs provide an overall estimate of an individual’s risk for different diseases [9]. Although individual variants typically have small to moderate effects on risk, their power increases when combined with a polygenic score. GWASs can find genetic information that can be used as a screening tool to identify individuals at risk, predict the risks of certain conditions, and diagnose diseases.
1. GWASs for type 2 DM
Several common genetic variants are associated with energy balance, appetite, insulin resistance, and insulin secretion [10]. Type 2 DM is a multifactorial disorder whose manifestation depends on multiple interacting environmental and genetic factors. Type 2 DM is a genetically heterogeneous disease, with several rare monogenic variants with large effects and a number of common variants with small to moderate effects involving complex interactions between genetic and environmental factors.
More than 400 loci for type 2 DM explain only 10% of its heritability, and all variants affect risk modestly, with a relative risk usually around 1.1−1 [2.11]. GWASs have identified that SNP (rs7903146) in TCF7L2 has the largest effect on risk, conferring a 1.4-fold increase per allele [12]. TCF7L2 is a Wnt signaling transcription factor that plays an important role in coordinating the expressions of proinsulin and insulin [12]. A risk variant of MTNR1B, encoding melatonin receptor 1B, modifies the RNA expression of MTNR1B by increasing enhancer binding for neurogenic differentiation factor 1 (NEUROD1) in human islets, which highlights the modulating role of melatonin in glucose homeostasis [13].
GWASs also have identified variants associated with obesity and a high body mass index (BMI). An SNP in the second intron of FTO was identified in GWASs for type 2 DM and obesity [14]. The association signal for type 2 DM disappeared completely with correction for BMI, indicating that the SNP increased type 2 DM risk by increasing BMI. A splice acceptor site variant that generates a premature stop codon and loss of function in ADCY3, which is highly expressed in visceral adipose tissues, was identified as the cause of the high risk of BMI and type 2 DM [15]. Functional studies in mice have suggested that ADCY3 may be a novel therapeutic target [15]. In addition, a combination of risk alleles, such as MNTR1B, G6PC2, and GCK, manifest fasting hyperglycemia and decreased insulin secretion [13], whereas those with variants in SLC30A8, HHEX, CDKAL1, CDKN2A/B, IGFBP2, and TCF7L2 show decreased insulin production and secretion [16].
GWASs conducted in various populations have revealed differences in the genetic risk factors that contribute to DM among different ancestral groups. A common SNP at a locus containing SLC16A11/13 in Latino, Mexican, and Japanese ancestry confers a 1.25-fold increased risk of DM; however, this has not been detected in populations of European ancestry [17,18]. Similarly, a common variant in TBC1D4 in individuals from Greenland strongly increases the risk of type 2 DM with an allele frequency of 17%, but this variant is extremely rare in continental Europe [19].
The genetics of type 2 diabetes (GoT2D) consortium collected comprehensive genome-wide sequence data from 2,657 cases of type 2 DM and controls in Europe. The Type 2 Diabetes Genetic Exploration by Next-Generation Sequencing in MultiEthnic Samples consortium focused on exome sequence variants from 12,940 individuals from 5 ancestry groups, including GoT2D exomes [10]. This study employed NGS to identify variants within the regions previously found in GWASs by genotyping arrays and imputation, suggesting that low-frequency variants contribute much less to the heritability of type 2 DM than common variants [10]. The UK Biobank transformed population research into a new phase with the announcement of exome sequences for 500,000 participants by 2019 [20]. The first report found that rare loss-of-function variants of FAM234A significantly reduced the risk of self-reported diabetes (odds ratio [OR], 0.64; 95% confidence interval [CI], 0.52–0.80; P=10-4). In addition, protein-altering variants in MAP3K15 are associated with lower serum glucose levels and protection against type 2 DM (OR, 0.85; 95% CI, 0.79–0.91, P=2.8×10-6) [20].
The generation of PRS can identify individuals’ future risk of DM, which can benefit from early intervention. A recent study of 18,197 cases and 423,697 controls from the UK Biobank showed that maximal discrimination (area under the curve C statistic of 66%) was obtained from a PRS of 136,795 variants [11]. These risk estimates are also conducted by the direct-toconsumer company 23andMe (https://www.23andme.com/), and they share results from 1,244 SNP-related PRS, with a recommendation of lifestyle interventions for high-risk customers [9].
The genes implicated in monogenic DM also contribute to polygenic forms; thus, they can be utilized as important therapeutic targets for prognosis prediction in type 2 DM [21]. Genes associated with monogenic DM, for instance, in patients with permanent neonatal diabetes caused by mutations in ABCC8 or KCNJ11 can be treated with high-dose sulfonylureas instead of insulin [21]. Individuals with maturity-onset diabetes of the young (MODY) types 1 and 3, caused by mutations in HNF4A and HNF1A, have shown the superiority of sulfonylureas over metformin or insulin [21]. The GLP1R gene encoding the receptor for glucagon-like peptide 1 (GLP-1) is a known to be associated with fasting glucose and type 2 DM [22]; this receptor is the target of GLP-1 analogs [22].
Collectively, the effect sizes and clinical significance are only small to modest; however, identifying risk loci provide a better understanding of the biological mechanisms of the disease and offers new therapeutic options.
2. GWASs for human height
GWAS results have demonstrated that height is highly polygenic, meaning that thousands of genetic variants contribute to an individual’s height. An early GWAS on adult height was performed of 4,921 individuals of European ancestry and identified a strong association between HMGA2 and height [23]. Although the genetic variant in HMGA2 explains approximately 0.4 cm of final height gain, this was the first study to demonstrate the genes relevant to growth through GWASs. Subsequently, the Genetic Investigation of Anthropometric Traits (GIANT) consortium conducted a GWAS in ~180,000 individuals of height. They reported that at least 180 loci influence adult height, and that these loci are not random but rather an enriched biological pathway for skeletal growth defects such as FGFR4, ECM2, and STAT2. They also found that 180 loci could explain, on average, 10.5% of adult height variance [24]. An expanded study of the GIANT Consortium found 697 variants clustered in 423 loci using approximately 250,000 samples of European ancestry, and they continued with more than 2 million samples with the release of the UK Biobank.25) This study indicated that human height is highly polygenic and that these SNPs explained more than 20% of its phenotypic variance [25].
Most GWASs conducted by the GIANT Consortium have been focused on European ancestry. GWASs have also been performed in Asian populations and identified 98 regions of the genome associated with height; 17 of these were unique to Asians [26]. In approximately 8,000 samples from Korea, 15 loci were associated with height, many of which had already been reported in the European population [27]. These findings suggest that most GWAS associations are shared across populations and that genetic regulation of height is similar globally.
A recent GWAS of 5.4 million individuals from diverse ancestries showed that 12,111 independent SNPs were significantly associated with height and accounted for 40% of the phenotypic variance in European populations [28]. This study demonstrated a strong genetic overlap of GWAS signals across ancestries; however, moderate SNP heterogeneity was observed between ancestries. Prediction results using GWAS revealed apparent attenuation in non-European ancestry, highlighting the need to further increase the sample size for GWAS in non-European populations.
GWASs are designed to map the polygenic architecture of common diseases; however, they have also benefited the detection of candidate genes of Mendelian growth disorders typically caused by rare genetic variants in a single gene. Variants of the same genes can have variable effect sizes and minor allele frequencies. For example, a highly infrequent variant of the Indian hedgehog (IHH) gene causes an extremely rare syndrome of acrocapitofemoral dysplasia. Individuals with acrocapitofemoral dysplasia carrying highly deleterious mutations in IHH are 2.3 to 8.6 standard deviation scores (SDSs) below the mean height. A rare missense variant in IHH is present in 0.2% of people and decreases height by 0.294 SDSs, while a common variant in IHH alters height by 0.05 SDSs per allele [29]. ACAN encodes the protein aggrecan, which is crucial for the structure and function of the growth palate and cartilage [30]. Homozygous mutations in ACAN cause a severe phenotype of spondyloepimetaphyseal dysplasia, whereas heterozygous mutations result in a milder phenotype of proportionate short stature in osteoarthritis. Recent GWAS results demonstrated that the most significant loci associated with height were observed on chromosome 15 near ACAN, and that multiple types of common variants in ACAN, including enhancers, missense variants, and tandem repeat polymorphisms, affect height [28]. Moreover, recent studies showed pathogenic variants in ACAN established as one of the common monogenic conditions in idiopathic short stature [30]. Such an overlap in regions highlighted by GWASs and Mendelian genetics illustrates an important aspect of the genetic architecture of height.
Using the UK Biobank-based GWAS of 253,299 European ancestries, PRS generated by 33,938 SNPs demonstrated the best predictive power [31]. Combined with age, sex, recruitment center, genotyping, and population stratification, PRS was able to capture 71.1% (95% CI, 70.8%–71.4%) of the total variance in adult height. This value was similar to the 72.6% (95% CI, 69.6%–75.6%) variance estimated using midparental height. When predicting adult short stature using sex and PRS, PRS achieved an area under the receiver operating characteristic curve (AUROC) of 0.843 (95% CI, 0.796–0.890) for identifying children who would have adult short stature, similar to the prediction by midparental height (AUROC, 0.879; 95% CI, 0.84–0.919). Furthermore, combining midparental height and PRS provided a better prediction accuracy than either metric alone. Therefore, the combination of traditional adult height predictors and genetic predictors enhances screening power for children at risk for short stature in adulthood.
3. GWASs for timing of puberty
Pubertal timing is a complex process influenced by genetic, nutritional, and environmental factors. The high correlation of pubertal timing within racial/ethnic groups and families, and between mono- and dizygotic twins suggests that this timing is regulated by genetic factors [32]. Age at pubertal initiation and menarche was reportedly younger in African-American than Caucasian girls [32,33]. Mothers’ age at menarche was associated with that of their daughters [34]. A constitutional delay of growth and puberty often has a familial component [35].
Studies using epidemiologic and intrafamilial tools suggest that 50%–95% of the variation in pubertal timing is determined by genetic control [32]. More than 100 genomic regions are associated with pubertal timing, and these genes are implicated in the hypothalamic-pituitary-ovarian axis involved in gonadotropin-releasing hormone (GnRH) secretion, pituitary development and function, hormone synthesis and bioactivity, energy homeostasis and growth, and peripheral feedback [32].
A GWAS for age at menarche performed in 2009 reported the first loci at LIN28B in the chromosome 6q21 region, which was reproducibly associated with population variations at pubertal timing [36]. LIN28 is an evolutionarily conserved RNA-binding protein that regulates mRNA translation and miRNA let-7 maturation at the onset of puberty; however, the detailed mechanism of puberty initiation remains unknown [36]. A recent GWAS of approximately 180,000 women of European descent found 106 genomic loci associated with age at menarche. Most variants showed very small effect sizes between 1 week and 5 months for each allele on the timing of menarche and significant enrichment of age at menarche-associated variants in rare puberty disorders, such as LEPR, TACR3, and GNRH1, as well as imprinted regions in a parent-of-origin-specific manner, such as DLK1, MKRN3, and KCNK9 [32]. In the largest study of 1,000 Genomes Project genomic data of 329,345 women of European ancestry, 389 independent genome-wide signals for age at menarche were identified [37]. These signals can explain approximately 7.4% of the variation in age at menarche, corresponding to approximately 25% of the estimated heritability [37]. This study also identified a significant role for imprinted genes in the regulation of pubertal timing with parent-of-origin–specific associations, such as MKRN3 and DLK1 [32]. In males, genetic studies of puberty are much fewer and smaller in scale because of lack of data on male pubertal milestones, however, a recent study using data from the UK Biobank and 23andMe reported that genes implicated in pubertal timing include ALX4, SRD5A2, and INHBB [38]. They also found a correlation between several adverse health outcomes and earlier male pubertal timing [38].
In the case of central precocious puberty (CPP), monogenic mutations in MKRN3, DLK1, KISS1, KISS1R, and other candidate genes, such as those biologically linked to gonadotropin signaling and those encoding steroidogenesis enzymes, have been reported [32]. A recent study characterized the genetic predisposition to CPP in girls of Han Chinese ancestry in Taiwan, and 105 loci were newly identified as genetic risk factors for early puberty [39]. They also found 33 SNPs from previous GWASs on pubertal timing [39]. Therefore, rare monogenic diseases and common SNPs during puberty share a genetic basis.
Early GWASs for age at menarche found significant signals in 4 loci previously associated with BMI, FTO, SEC16B, TRA2B, and TMEM18, and 3 in or near genes implicated in energy homeostasis, BSX, CRTC1, and MCHR2 [40]. Similarly, in GWASs that expanded the sample size and number of identified loci, several loci, including pubertal timing and BMI, overlapped [37,39]. Both energy balance and reproduction are modulated by peripheral signals, such as leptin and ghrelin [40].
Founder effects in pediatric endocrine diseases
1. Founder effects and genetic drift
GWASs in isolated populations is helpful for identifying the founder effect due to geographic or cultural barriers that have isolated genetic flows in neighboring populations. Founder mutations can result from a true founder event or a bottleneck effect (Fig. 2). The former occurs when a subset of individuals is separated from a larger population and establishes a new population, while the latter is caused by a marked reduction in population size due to migration or isolation causing changes in allele frequency [41]. Founder populations can increase the frequency of certain autosomal recessively inherited Mendelian disorders through genetic drift. Genetic drift causes random fluctuations in the number of alleles in a population, resulting in a dramatic reduction in the population size before recovery (bottleneck effect). Therefore, populations with strong genetic drift tended to have one predominant mutant allele. The allele frequencies of common and rare genetic variants in a population are influenced by natural selection through evolution and de mographic history [41].

Effects of genetic drift and evolutionary force in an isolated population. Marked reductions in population size due to migration or isolation cause changes in allele frequency, followed by reduced diversity and founder effects.
The “Out of Africa” theory suggests that modern humans originated from a small population residing in Africa [42]. Accordingly, the most common genetic variants can be traced back to the ancient African population, from whence they are shared across the world, whereas rare variants are typically restricted to closely related populations [42]. A great demographic expansion began approximately 45,000–60,000 years ago in Africa, when a highly rapid migration of populations occurred across the Eurasian continents [35]. Accordingly, there is a continuous loss of genetic diversity within populations living outside Africa, resulting in “serial founder effects.”
2. Founder mutations in endocrine diseases
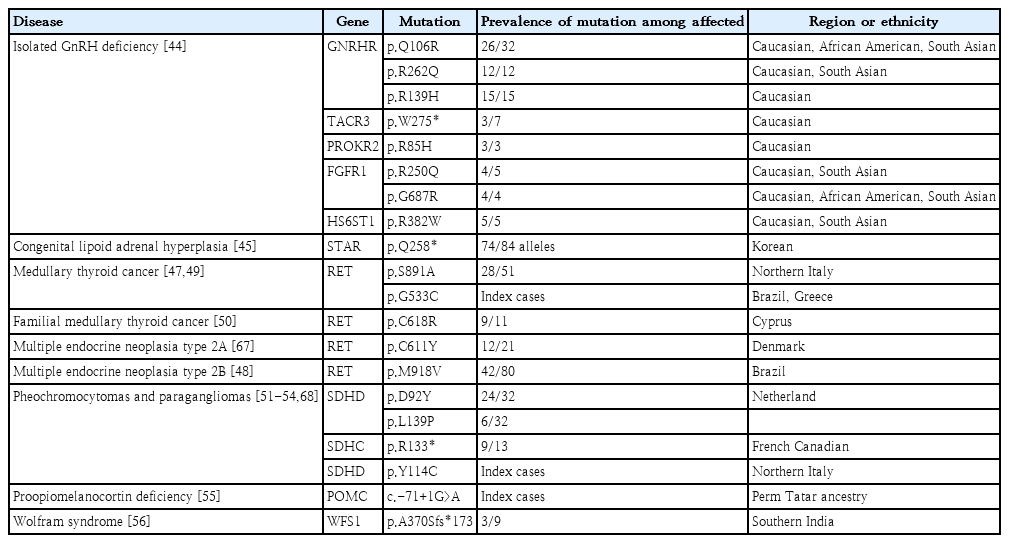
Several founder mutations have been identified in endocrine disease (Table 1). Isolated GnRH deficiency (IGD) is a rare reproductive disorder with remarkable allelic and locus heterogeneity. Over the last 3 decades, genetic approaches have identified >60 IGD genes with various modes of inheritance [43]. The typical phenotype of IGD is the consequence of loss-of-function mutations, most of which are private and nonrecurrent [44]. However, haplotype analyses revealed 8 mutations in 5 genes, including GNRHR, TACR3, PROKR2, FGFR1, and HS6ST1 founder mutations [44]. The estimated age of the 4 mutant alleles in GNRHR, TACR3, FGFR1, and HS6ST1 was approximately 5,000 years, corresponding to a time of rapid population expansion. In contrast, the PROKR2 p.L173R founder mutation is approximately 9,000 years old and may confer a heterozygote advantage. This finding suggests that the persistence of the lossof-function alleles in diverse populations might reflect a potential heterozygote advantage for carriers.
Founder mutations in endocrine diseases
As the Korean population has settled in the Korean peninsula as a single nation for thousands of years, several recurrent mutations have been identified as founder mutations. The p.Q258* mutation of STAR in congenital lipoid adrenal hyperplasia is another founder mutation [45]. This mutation is relatively common in China, Japan, and Korea. These countries are geographically adjacent and culturally influenced by people’s movement. The age of the mutation is estimated at approximately 5,000 years, corresponding to the time at which the Korean people settled on the Korean peninsula. Therefore, studying the founder effect can provide information about a population’s evolution and migration pathways, thus enabling the screening of at-risk individuals.
Pathogenic variants of RET cause multiple endocrine neoplasia type 2 (MEN2), an autosomal dominantly inherited cancer syndrome. Codon-specific pathogenic variants in RET are strongly correlated with the MEN2 phenotype [46]. Cysteine-rich extracellular domains in exons 10 and 11 frequently undergo genetic alterations and are associated with pheochromocytoma, primary hyperparathyroidism, or both. In a recent comprehensive study of the geographical profile of RET variants, codon 634 mutations seemed to be the most prevalent worldwide; however, founder effects of uncommon variants have been observed in some countries [46-50]. Other cancer-predisposing genes encoding succinate dehydrogenase (SDH) subunits cause paragangliomas (PGL). National datasets have identified a high prevalence of particular pathogenic variants in SDHx according to geographical region [51,52]. The SDHD p.Y114C variant in the Mocheni valley close to Trentino, Italy, probably originated from Germany 600–700 years ago [52]. The SDHC p.R133* mutation has been reported in PGL cases among French Canadians [53]. In the Netherlands, the SDHD founder mutation p.D92Y, accounted for almost 70% of all SDHx carriers/cases [54]. The Dutch founder effect for PGL was also noted in South Africa and is potentially derived from historical Dutch emigration [51]. This molecular epidemiology can be helpful in screening for genetic risks and preventing the occurrence of cancer in atrisk individuals.
Seven unrelated patients of the Perm Tatar ethnic group presented with hypoglycemia, excessive weight gain, and low plasma adrenocorticotropin and cortisol levels [55]. These patients had unusual clinical features of chronic obstructive pulmonary disease. Using whole-exome sequencing (WES) and functional assays, a homozygous variant of the 5'-untranslated region in POMC (c.-71+1G>A) led to a significant decrease in POMC mRNA expression, and haplotype analyses suggested a founder effect, which occurred at least 106.7 years ago. Another rare monogenic form of diabetes, Wolfram syndrome type 1, is reportedly associated with the founder effect of WFS1 in Southern India [56]. This study identified 11 patients carrying WFS1 variants using NGS and identified a recurrent WFS1 mutation p. A370Sfs*173. Based on haplotype analysis, microsatellite markers were shared among patients harboring the mutation. These rare disorders can lead to severe comorbidities; therefore, early diagnosis and genetic counseling are crucial.
Founder mutations have generated considerable interest in human genetics because their study may facilitate tracing the ancestry, migration, and history of human populations. Screening for one or a few prevalent founder mutations is more efficient than testing for many rare mutations. Additional advantages of founder mutation screening are identifying simple ways to identify at-risk groups, developing new ideas for preventing and treating conditions associated with these mutations, and studying their prevalence and penetrance in the population.
NGS for pediatric endocrine diseases
1. Development of NGS
Testing a single or small number of genes may be preferable if a disease is highly phenotypically and/or genetically homogeneous. However, NGS can also be applied to phenotypically and genetically heterogeneous conditions. Its availability allows both the identification of novel candidate genes and an in-depth understanding of the architecture of several endocrine diseases. Several different NGS approaches are available that allow the sequencing of several regions of interest or the entire exome or genome (Fig. 3).
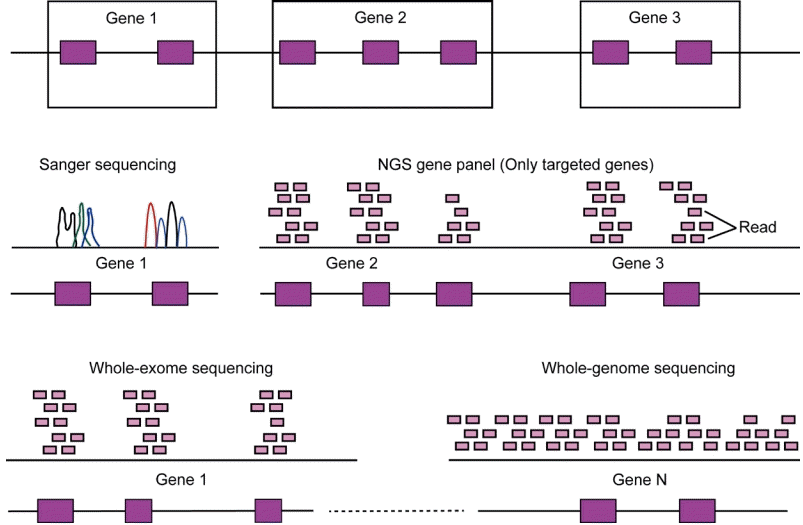
Comparison of sequencing platforms. Compared to Sanger sequencing, targeted gene panel or whole-exome sequencing utilize sequence reads concentrated over the coding portions of genes. In contrast, whole-genome sequencing analyzes almost the entire genomic sequence. NGS, next-generation sequencing.
NGS is another genomic approach for the treatment of pediatric endocrine disorders. NGS platforms perform massive parallel sequencing of multiple genes, entire exomes, and genomes. NGS is efficient and cost-effective compared with sequential gene testing using Sanger sequencing [2]. The diagnostic efficiency of clinical WES for rare Mendelian disorders is 20%–30% [57]. However, in patients with distinctive phenotypes, such as neonatal diabetes or primary adrenal insufficiency, the diagnostic yield increases to 80% [58,59]. Therefore, optimal sequencing platforms must be selected according to the patient phenotype and genetic heterogeneity. Testing for a single or small number of targeted genes may be preferable if the disease is phenotypically and/or genetically homogeneous. However, whole-exome or genome sequencing can be used under phenotypically and genetically heterogeneous conditions.
2. Applications of NGS for pediatric endocrine diseases
The first discovery of gene mutations in endocrine diseases using NGS was published in 2011 [60]. This study identified 2 recurrent somatic mutations in KCNJ5 in eight of 22 patients with aldosterone-producing adenomas. The patient presented with hypertension and primary hyperaldosteronism. An in vitro study demonstrated increased sodium conductance through mutant channels leading to membrane depolarization of adrenal cortical cells and stimulation of aldosterone release and cell proliferation [60].
In this context, NGS can be applied to many pediatric endocrine disorders with genetic heterogeneity, such as hypogonadotropic hypogonadism, disorders of sex development (DSD), skeletal dysplasia, Noonan syndrome and related disorders, congenital pituitary hormone deficiency, and monogenic diabetes (Table 2). Loss-of-function mutations in 2 causative genes, MKRN3 and DLK1, were identified using NGS in patients with CPP. MKRN3 plays a role in the inhibition of factors that stimulate pubertal pulsatile GnRH secretion, while DLK1 acts as an adipogenesis gatekeeper by preventing adipocyte differentiation. A recent meta-analysis suggested that mutations in MKRN3 occur more frequently found in familial cases (prevalence 19%; 95% CI, 0.05–0.36) than in sporadic cases (prevalence, 2%; 95% CI, 0.01–0.04) [61]. A subgroup analysis showed that the prevalence was higher in males, familial cases, and non-Asian countries [61]. Although loss-of-function mutations in DLK1 and KISS1 cause CPP, they have rarely been reported [32]. Interestingly, MKRN3 and DLK1 are both maternally imprinted and paternally expressed genes [32]. These findings suggest that genomic imprinting plays a role in regulating the timing of human puberty.
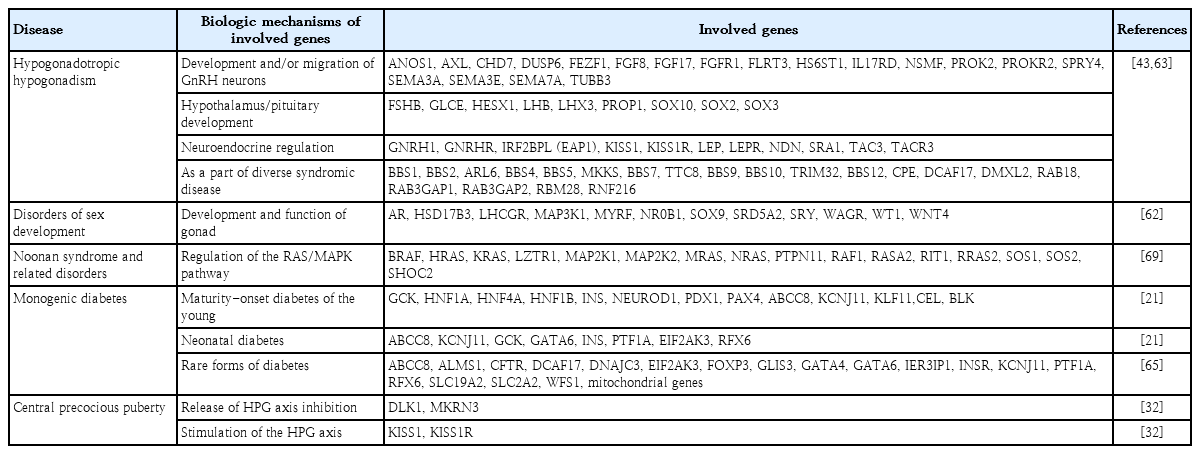
Applications of next-generation sequencing for genetic diagnosis of pediatric endocrine diseases
DSD is caused by several genetic etiologies with varying phenotypes, and genetic diagnosis using Sanger sequencing can be achieved in only 20% of cases. The diagnostic efficiency of targeted gene panel sequencing of 67 genes was approximately 30% in 44 patients with DSD [62]. NGS can be considered a firsttier diagnostic tool for DSD with diverse genetic heterogeneity and a wide phenotypic spectrum. However, the diagnostic yield is no more than 50% because of key novel genes and genomic changes in enhancers or regulatory regions, copy-number variants, somatic changes during early embryonic life, or epigenetic or environmental factors [2], necessitating whole-genome sequencing or mapping technologies.
IGD is another endocrine disorder with genetic heterogeneity. In our group, the targeted gene panel sequencing of 69 genes identified pathogenic or likely pathogenic variants in 37% of patients [63,64]. Mutations in the FGFR1 gene were the most common [63]. However, genetic defects in IGD cannot be identified in over 50% of cases because of technical limitations of WES, mutations in noncoding areas, or cryptic, structural defects in the genome, or epigenetic factors.
Monogenic diabetes is characterized by a Mendelian inheritance pattern with a large effect size of causal variants and minimal environmental contribution.21) Molecular diagnosis using NGS is also useful for monogenic diabetes as described previously [21]. Genetically confirmed monogenic diabetes accounted for 5.1% of patients in a single-center study in Korea [65]. In targeted gene panel sequencing of 109 Korean patients with suspected monogenic DM, 21% of the patients harbored pathogenic or likely pathogenic variants [66]. The characteristics of patients with pathogenic or likely pathogenic variants include a lower BMI, higher MODY probability, and lower C-peptide levels [21]. Molecular genetic approaches for monogenic DM have clinical implications, including providing therapeutic strategies, such as insulin or sulfonylureas, and predicting outcomes such as transient or permanent DM [21].
Conclusions
Recent advances in molecular genetics have advanced our understanding of the molecular mechanisms of pediatric endocrine disorders and play a role in mainstream medical practice. The improved predictive power of GWAS variants makes the application of GWAS data a useful clinical tool. The identification of founder effects within a population offers the powerful advantages of efficiently localizing genes that underlie Mendelian disorders. NGS technology is beneficial for clinical practice and research on pediatric endocrinology. In addition, genomewide sequencing offers the benefit of reanalysis over time to incorporate advances in knowledge. In summary, the application of molecular technology has allowed us to further our understanding of endocrine diseases and has an impact on the diagnosis and genetic counseling of pediatric endocrinology.
Notes
Conflicts of interest
No potential conflicts of interest relevant to this article are reported.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (Ministry of Science and ICT) (No. NRF2021R1F1 A104593011).