Global prevalence of classic phenylketonuria based on Neonatal Screening Program Data: systematic review and meta-analysis
Article information

Abstract
Phenylketonuria is a disease caused by congenital defects in phenylalanine metabolism that leads to irreversible nerve cell damage. However, its detection in the early days of life can reduce its severity. Thus, many countries have started disease screening programs for neonates. The present study aimed to determine the worldwide prevalence of classic phenylketonuria using the data of neonatal screening studies. The PubMed, Web of Sciences, Sciences Direct, ProQuest, and Scopus databases were searched for related articles. Article quality was evaluated using the Joanna Briggs Institute Critical Appraisal Evaluation Checklist. A random effect was used to calculate the pooled prevalence, and a phenylketonuria prevalence per 100,000 neonates was reported. A total of 53 studies with 119,152,905 participants conducted in 1964–2017 were included in this systematic review. The highest prevalence (38.13) was reported in Turkey, while the lowest (0.3) in Thailand. A total of 46 studies were entered into the meta-analysis for pooled prevalence estimation. The overall worldwide prevalence of the disease is 6.002 per 100,000 neonates (95% confidence interval, 5.07–6.93). The metaregression test showed high heterogeneity in the worldwide disease prevalence (I2=99%). Heterogeneity in the worldwide prevalence of phenylketonuria is high, possibly due to differences in factors affecting the disease, such as consanguineous marriages and genetic reserves in different countries, study performance, diagnostic tests, cutoff points, and sample size.
Key message
Question: What is the global prevalence of classic phenylketonuria based on Neonatal Screening Program Data?
Finding: The overall worldwide prevalence of the disease is 6.002 per 100,000 neonates. The highest prevalence (38.13) was reported in Turkey, while the lowest (0.3) in Thailand.
Meaning: This difference in the prevalence may be due to differences in the number of consanguineous marriages among the different regions, phenylalanine cutoff points, and sample sizes.
Graphical Abstract

Introduction
Genetic and congenital abnormalities are the most important causes of death and malformation in the first month of life [1]. Phenylketonuria (PKU) is an inborn error of amino acid metabolism caused by phenylalanine hydroxylase gene mutations [2,3]. PKU patients experience an irreversible decrease in intelligence quotient scores, suppressed verbal function, impaired attention, and underdeveloped motor control skills [4,5].
The early diagnosis of PKU before the end of the first month of life is critical to controlling hyperphenylalaninemia [4,6]. Children with PKU seem normal during the first days of life; however, nervous system damage progresses gradually and becomes apparent over several months [7]. The early detection of PKU in the asymptomatic period and treatment with a phenylalanine restricted diet is warranted to ensure normal development [8-12]. Therefore, neonatal screening as a fundamental public health intervention started in the mid-20th century [4,12,13].
Since PKU has autosomal recessive inheritance, consanguineous marriage is an important risk factor [1,4]; thus, countries with a high prevalence of consanguineous marriages have high disease prevalence [6,14]. PKU varies among ethnic groups, races, and geographic regions. For example, In Japan, the incidence is reportedly 1:108,822 [15]. Turkey, with an incidence of 1:6,000, and Iran, with an incidence of 1:4,698, are among the countries with the highest PKU incidences [16,17].
Despite numerous studies conducted in various countries on PKU prevalence using screening programs, no study has systematically compared the prevalence of PKU across regions and countries or sources of heterogeneity. To address this gap, this systematic review and meta-analysis aimed to investigate the worldwide prevalence of PKU. Moreover, many countries have acknowledged the benefits of newborn screening programs for PKU. Moreover, newborn screening programs have enabled the rapid and large-scale testing of many children with good quality control.
Methods
This systematic review adhered to the guidelines of the Joanna Briggs Institute Reviewers’ Manual 2014, Systematic Review of Prevalence Data [18]. The PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) 2009 flow diagram was used to guide the study identification and selection process [19].
1. Search method
The PubMed, Web of Sciences, Sciences Direct, ProQuest, and Scopus databases were searched on Oct 28, 2018, without publication date restrictions. The search strategy carefully captured all potentially eligible records of PKU prevalence. A combination of medical subject headings (MeSH) and similar text words in English was used. Google Scholar was searched, as were the reference lists of the reviewed articles to identify additional relevant articles.
The key MeSH terms were as follows: (Infants OR Newborns OR Neonate) AND (Phenylketonuria OR Hyperphenylalaninemia, Non-Phenylketonuric OR BH4 Deficiency OR Tetrahydrobiopterin Deficiency OR Phenylketonuria II OR DHPR Deficiency OR Dihydropteridine Reductase Deficiency OR Atypical PKU) AND (Incidence OR Prevalence) and Screening. No time limitation was considered for the database search.
2. Inclusion criteria
All original articles that directly reported PKU prevalence based on newborn screening of populations were included. A newborn, infant, or neonate is a child younger than 28 days of age; in this review, the sampling period was limited to the first 28 days of life. All studies were included if they used a laboratory screening test for disease detection.
Reviews, comments, and letters were excluded. Moreover, studies that reported the prevalence in a selective neonatal population (congenital diseases, intellectual disability), those that included children older than 28 days, and those that indirectly estimated prevalence according to consanguinity or the incidence of another genetic disease were excluded. In some cases, studies were conducted of the same PKU prevalence in one country using different dates; in such cases, the more recent study (which also included the data from the older study) was included and the older study was omitted. Moreover, studies that detected PKU based on clinical manifestations in neonates or neural tube defects in fetuses were excluded.
3. Data collection
The title, abstract, and keywords of every identified article were carefully scanned and relevant articles were selected by title or abstract review.
4. Data extraction and management
Two reviewers (HSH. and FZ) independently extracted the patient characteristics, study characteristics, screening test used, and incidence from the reviewed studies using a data extraction form. Any disagreements between the 2 researchers were solved by consultation of another reviewer or a clinical adviser. Abstracts not published as full texts were not included in the study. To avoid data entry errors, all results were double-entered into a data extraction form. The included studies used different units such as mmol/L, μmol/dL, and mg/dL to report phenylalanine level. However, in this study, to increase comparability, all units were converted to mg/dL. In addition, to ensure more accurate comparisons, PKU prevalence was calculated as percentage and rate per 100,000 screened neonates.
5. Assessment of methodological quality
Article quality was assessed using the Joanna Briggs Institute Critical Appraisal Checklist for studies that reported prevalence data. Each article was evaluated according to the following methodological criteria: appropriate sample, adequate sample size, valid methodology, valid measure to detect the disease, and an appropriate statistical analysis.
6. Risk of bias
Risk of bias was assessed using the risk of bias tool for studies measuring disease prevalence designed and developed by Hoy et al. [20] Based on this 10-point checklist, studies were assessed for internal and external validity and grouped as having high, moderate, or low bias risk. Studies with a score of 9–10 were considered at having low risk of bias; 6–8, as having moderate risk; and less than 6, as having high risk. Those studies with a high risk of bias were excluded from the meta-analysis.
7. Data analysis
The data were analyzed using Stata ver. 12 (StataCorp LP., College Station, TX, USA). In a meta-analysis, pooled prevalence was estimated based on World Health Organization (WHO) regions and reported as per 100,000 neonates/population with 95% confidence interval (CI).
The degrees of heterogeneity among the included studies are expressed by the I2 heterogeneity statistic, and the random effects model was used to estimate the pooled prevalence in subgroups. A forest plot was used to display the meta-analysis results.
The mixed model test considered WHO regions as a random intercept. In this test, phenylalanine levels were modeled as independent variables, while prevalence was considered a dependent variable.
Furthermore, a meta-regression analysis was performed to investigate the impact of variables such as WHO region, phenylalanine cutoff point, study period, national or governmental screening program, and participant age on the I2 and pooled prevalence.
Results
1. Study selection
After a comprehensive search, 1,228 relevant articles were identified, 608 duplicates were removed. The relevance of the remaining 1,346 studies was evaluated based on the titles/abstracts alone; of them, 99 studies were subjected to fulltext review, which eliminated another 46 studies according to the inclusion and exclusion criteria. Finally, 53 studies with 119,152,905 participants were included in the present systematic review; of them, 46 were included in the meta-analysis (Fig. 1).
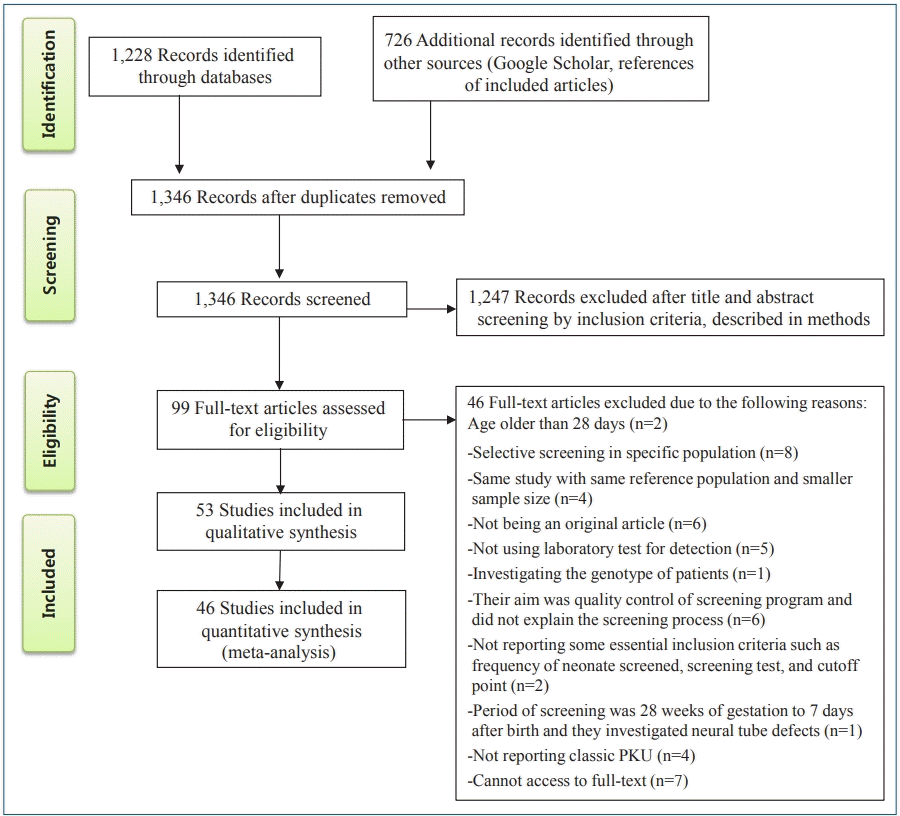
Flow diagram of the literature search and study selection process. PKU, phenylketonuria.
2. Risk of bias
In the bias assessment, 7 studies scored below 6 (high risk) and were excluded from the meta-analysis; thus, the pooled global prevalence was estimated without them [11-13,21-24]. Moreover, 33 of the studies scored 6–8 (moderate risk) [1,9,16,17,25-53], while the other 13 studies had low risk [2,10,54-64].
3. Study characteristics
In this section, the systematic review results are presented based on different characteristics, including region, test, and participant characteristics.
4. Region characteristics
The included studies (1964–2017) are presented in Table 1. The longest study, conducted in France, examined 35 years of screening data from 1966 to 2001 [56]. The largest screening population was in China (35,795,550 newborns for 30 years from the start of the screening project); the largest number of cases was detected (3,082 patients) in this study [60]. However, the smallest population belonged to a study conducted in Iraq in 2015, and only 8,255 newborns were screened [13].
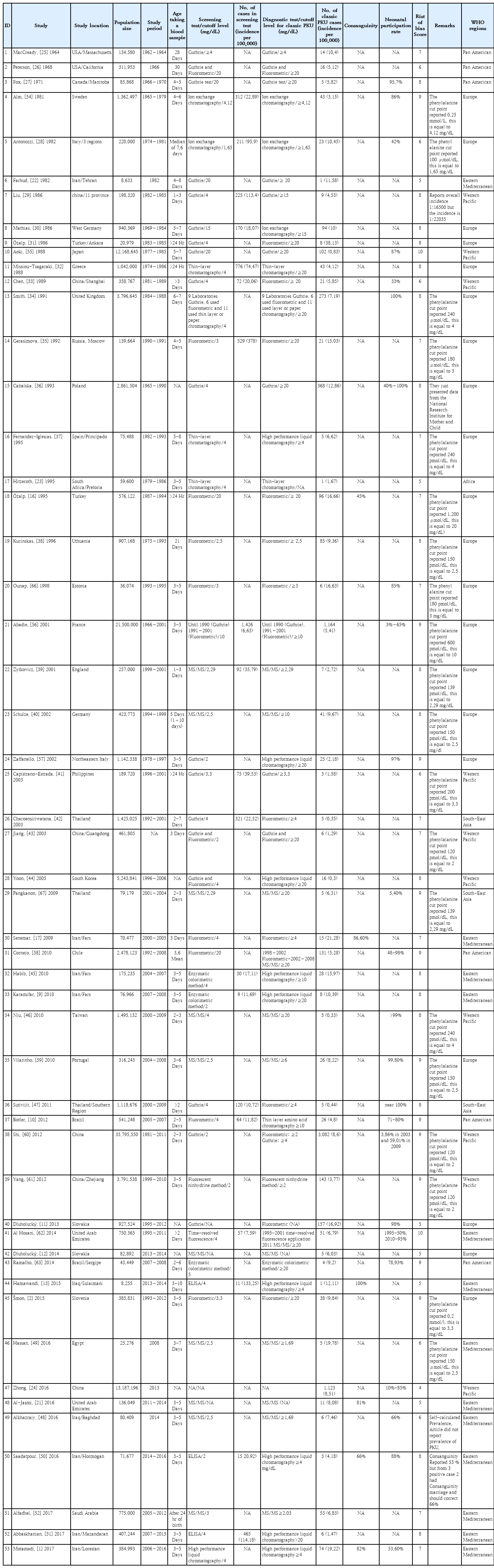
Description of studies included in the study
5. Participants’ characteristics
Sampling age at screening was below 5 days in 30 studies [1,2,9,10,16,17,21,23,27,29,31-33,35,39,41,43-46,48,50-53,56-58,60,61], 5–10 days in 15 studies [13,22,28,30,34,37,42,47,49,54,55,59,62,63,65], and over 10 days in 3 studies [25,26,38]. Five studies did not report newborn age at screening [11,12,24,36,64].
The neonatal participation rate was reported in 23 studies [1,10,11,24,27,28,33,34,44,46-48,50,53-60,62,63]; of them, it was above 90% in 7 studies [11,27,34,46,47,57,59]. The participation rate increased with the progression of the screening process in 8 studies [1,24,27,36,56,58,60,62].
Moreover, 6 studies reported the percentage of consanguineous marriages among the parents of newborns with PKU [1,13,16,17,21,50]. The percentage of consanguineous marriages varied from 45% in Turkey [16] to 100% in Iraq [13].
6. Test characteristics
In the included studies, 2 stages were used to diagnose infants with classical PKU.
7. Screening tests
A total of 19 studies reported the number of positive cases in the first stage of screening. The phenylalanine cutoff point for separating positive cases and referrals for diagnostic testing ranged from 1.65 mg/dL to 20 mg/dL. The highest recall rate in the first stage of screening was 378 per 100,000 neonates in a study conducted in Russia [35].
8. Diagnostic tests
In the diagnostic stage, the phenylalanine cutoff point for diagnosing classic PKU patients ranged from 1.65 mg/dL to 20 mg/dL. Moreover, 22 studies selected 20 mg/dL as the positive cutoff point and 5 studies did not report a cutoff point [2,9,16,22,25,26,31-36,43,44,46,51,55,57,58,62-64].
9. Pooled global prevalence of classic PKU
Among the included studies, the highest prevalence was found in Turkey (38.13), followed by Iran, with a prevalence of 21.28 per 100,000 neonates [17,31], while the lowest prevalence was reported in studies conducted in Thailand (0.3) and Taiwan (0.44) [42,46 47,64].
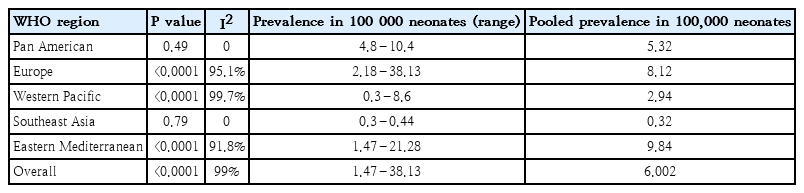
A subgroup estimation of the polled prevalence showed that the pooled prevalence of classic PKU in the included studies was 6.002 (95% CI, 5.07–6.93). The highest prevalence was seen in Eastern Mediterranean (9.83; 95% CI, 6.18–13.48), Europe (8.11; 95% CI, 6.54–9.69), Pan America (5.32; 95% CI, 4.47–6.07), Western Pacific (2.94; 95% CI, 0.91–4.97), and Southeast Asia (0.32; 95% CI, 0.19–0.45) per 100,000 neonates (Fig. 2).

Forest plot of pooled global prevalence of phenylketonuria. ES, estimated; CI, confidence interval.
10. Statistical analysis
According to the results of mixed model, cutoff point selection had no effect on prevalence. The p value obtained from the likelihood ratio test in the mixed model test suggested that the random intercept model was appropriate. Moreover, based on the intraclass coefficient, 29% of the PKU prevalence changes in different countries were justified by consideration of the WHO regions (Table 2).
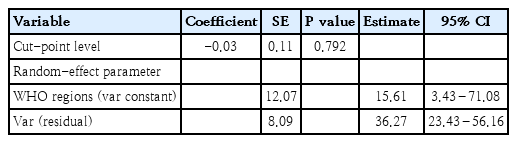
Result of mixed model test
A meta-regression test was used to assess the effect of year, phenylalanine cutoff point, region, neonate age at screening, and screening level (national or regional) on heterogeneity.
In the naïve model without any variables, I2 was 99%. Several models with different variables were created in which I2 ranged was 97%–99%, and the input of different variables did not decrease the heterogeneity.
Among the variables included in the model, only some WHO regions were significant. In the meta-regression model, the European region was selected as a reference. In the Eastern Mediterranean region, the prevalence was 1.01 greater than that in the European region, but the difference was not significant. The pooled prevalence of the different regions is reported in Table 3.
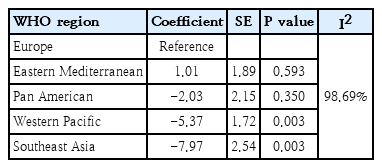
Result of meta-regression test
According to I2 by region and overall, the studies had high heterogeneity (Table 4).
Prevalence rate and heterogeneity in regions
Discussion
This systematic review aimed to investigate the worldwide prevalence of PKU and provided a general picture of its status. The results of this study demonstrate that the worldwide prevalence of the disease is 0.3–38.13 per 100,000 newborns. However, the meta-analysis revealed that the I2 index, which indicates heterogeneity, was reported for all regions except Southeast Asia (91.8%) and Pan America (99.7%), indicating high heterogeneity among countries and regions.
The uni- and multivariate models in the meta-regression showed that phenylalanine level, geographical area, neonate age at screening, screening level (national or regional), after the control for year of study, did not change heterogeneity.
However, the differences in prevalence can be attributed to 2 factors: (1) variability of the factors affecting disease worldwide; and (2) differences in the methods used in the studies.
PKU is a c0ngenital genetic disease; thus, factors such as culture, customs, consanguineous marriage, and genetics are expected to affect its incidence but among included studies only 6 studies in Iran, Iraq, Turkey, and the United Arab Emirates [1,13, 16,17,21,50] reported consanguineous marriages among parents of children with PKU.
Therefore, a lack of information about the prevalence of PKU in many countries in which consanguineous marriage is prevalent and a lack of reporting consanguineous marriage status in parents of children with PKU in many studies prevented us from controlling the effect of this variable on prevalence.
The next important determinant of prevalence is study performance; factors such as diagnostic tests, cutoff point, and sample size can affect the pooled prevalence in prevalence studies.
However, in the mixed model test, there was no significant relationship between cutoff point and disease prevalence, which might have been due to the effect of the confounding variables. However, the difference was noticeable when the cutoff point differed in the same population and within the same country. For example, in 3 studies conducted during 2000–2008 in Fars province (Iran), a different cutoff point was found. Moreover, Senemar chose a phenylalanine level of ≤4 mg/dL to define classical PKU and reported a prevalence of 21.28 [17]. Habib et al. [45] considered a phenylalanine ≤10 mg/dL cutoff value and reported a prevalence of 15.97. Furthermore, in the study of Karamifar et al. [9], phenylalanine levels above 20 mg/dL were considered positive and a prevalence of 10.39 was reported.
Sample size is the other factor involved in the difference in prevalence among studies. In a meta-analysis, the pooled prevalence is estimated according to the sample size, and larger studies have greater impact on prevalence. Thus, although studies conducted in the Eastern Mediterranean region reported higher prevalence than those in the Western Pacific region, since most studies in the latter had a larger sample size and the weighted sample size in that region was 24.83%, higher than that in the Eastern Mediterranean region (16.2%), the pooled prevalence in studies conducted in the Eastern Mediterranean region was close to that of the Western Pacific region.
Although this study addressed an important concern in genetic diseases, its findings may not be highly accurate, as there were many sources of heterogeneity in the reviewed studies that could have affected their pooled prevalence. Moreover, some heterogeneous sources might not have been identified. However, the standardization of study methods can partly solve this problem.
One of the limitations of this study was the failure to report consanguineous marriage in parents of newborns with PKU. Thus, it was not possible to answer the following question:
Is the difference in PKU prevalence among different countries due to differences in the number of consanguineous marriages?
Thus, we suggest that consanguineous marriages be recorded and reported in screening programs designed to identify patients with PKU and other congenital metabolic diseases.
In conclusion, all relevant studies conducted in 1964–2017 were included in this review. The highest PKU prevalence was observed in Turkey (38.13), while the lowest was seen in Thailand (0.3). Among the WHO regions, the highest prevalence belonged to Eastern Mediterranean Regional Office, while the lowest was in Southeast Asia. This difference in the prevalence may be due to differences in the number of consanguineous marriages among the different regions, phenylalanine cutoff points, and sample sizes.
Notes
No potential conflicts of interest relevant to this article are reported.
Acknowledgements
The authors thank Moslem Taheri for his advice for extraction, Razieh Zahedi for her advice for the meta-analysis, Maryam Nazemzadeh for professional English editing.