Autoimmune encephalitis and epilepsy: evolving definition and clinical spectrum
Article information

Abstract
Advances in autoimmune encephalitis studies in the past 10 years have led to the identification of new syndromes and biomarkers that have transformed the diagnostic approach to the disorder. The disorder or syndrome has been linked to a wide variety of pathologic processes associated with the neuron-specific autoantibodies targeting intracellular and plasma membrane antigens. However, current criteria for autoimmune encephalitis are quite dependent on antibody testing and responses to immunotherapy, which might delay the diagnosis. This form of encephalitis can involve the multifaceted presentation of seizures and unexpected behavioral changes. The spectrum of neuropsychiatric symptoms in children is less definitive than that in adults, and the incorporation of clinical, immunological, electrophysiological, and neuroradiological results is critical to the diagnostic approach. In this review, we document the clinical and immunologic characteristics of autoimmune encephalitis known to date, with the goal of helping clinicians in differential diagnosis and to provide prompt and effective treatment.
Introduction
Acute encephalitis is an inflammatory condition of the brain with multiple etiologies that has a crucial impact on relevant morbidity, occurring across all ages, although more common in children [1]. Encephalitis etiology remains unknown in >50% cases. This explains why the criteria for encephalitis diagnosis are mainly based on infection characteristics. The Consensus Statement of the International Encephalitis Consortium reported that an encephalitis diagnosis required evidence of an altered mental status lasting ≥24 hours with no alternative cause identified (major criterion) along with ≥2 minor criteria, including fever ≥38°C (72 hours before or after presentation), generalized or partial seizures not fully attributable to a pre-existing seizure disorder, new-onset focal neurologic findings, cerebrospinal fluid (CSF) pleocytosis (white blood cell [WBC] count ≥5/mm3), abnormal brain parenchyma on neuroimaging, and abnormalities on electroencephalography (EEG) not ascribable to other causes [1]. Two of these minor criteria were required for a possible diagnosis of encephalitis, while ≥3 are needed for a probable or confirmed diagnosis.
However, the possible role of autoimmunity in encephalitis development has been reported in the last decade [2]. The most inspiring progress came with the discovery of multiple neural-specific autoantibodies in patients with acute seizures and status epilepticus (SE) intractable to antiepileptic drug (AED) therapy. Increasing evidence supports an autoimmune etiology for seizures in the absence of these syndromic manifestations (e.g., limbic or extralimbic encephalitis) for a patient subgroup showing drug-resistant epilepsy [3,4]. Clinical hints include acute or subacute onset (over days to weeks), a remarkably high seizure frequency, seizure variability or multifocality, AED resistance, personal or family history of autoimmunity, and recent or past neoplasm history [3,4]. Rapidly evolving cognitive impairment, neuropsychiatric symptoms, evidence of multilevel central nervous system (CNS) involvement, or new-onset movement disorder propose but are not necessary for diagnosis.
Cases with an autoimmune pathogenesis have a wide spectrum of clinical manifestations; some features differ significantly from those that are usually found in infectious encephalitis. We aimed to report a case of confirmed anti-N-methyl-D-aspartate (NMDA) receptor encephalitis and to review the immunologic and clinical characteristics of autoimmune encephalitis to support clinicians with differential diagnosis and providing early effective treatment.
Case
A developmentally appropriate and previously healthy 15-year-old girl presented to the Emergency Department with a 4-day history of severe and persistent headache associated with a 1-day history of photophobia, altered mentality, and gait disturbance. No recent contributing history was noticed. She was afebrile but had tachycardia (111 beats/min) and hypertension (146/78 mmHg). She was 160 cm tall and weighed 51 kg. The initial neurologic examination revealed lethargic mentality with emotional lability, dysarthria, and an ataxic gait without lower-extremity weakness; however, no focal neurologic deficits or meningismus-related signs were apparent. She had no palpable lymphadenopathy or rash.
The initial work-up showed normal complete blood count, electrolyte, and chemistry panel, and thyroid function test findings. Pregnancy testing was negative. Brain magnetic resonance imaging (MRI) showed unremarkable results. Lumbar puncture revealed significant lymphocytic pleocytosis (WBC, 114/mm3; 86% lymphocytes); normal protein (21 mg/dL) and glucose (64 mg/dL) levels; and normal opening pressure. CSF meningitis and encephalitis panels were negative for multiple bacterial, viral, and fugal antigens, including herpes simplex virus (HSV)-1, varicella zoster virus, and Cryptococcus. Systemic infectious and rheumatologic evaluations were also negative. Pelvic computed tomography of a palpable mass found on an abdominal exam revealed a large cystic mass with a complex component (3.8 cm×4.9 cm×3.6 cm) in her right ovary, which was most likely a dermoid cyst or teratoma (Fig. 1). She underwent surgical resection of the ovarian tumor. Twenty-four-hour video/EEG monitoring revealed continuous diffuse background slowing and extreme delta brushes (Fig. 2), suggesting diffuse encephalopathy related to anti-NMDA receptor (NMDAR) encephalitis. No AED was administered. The antibody and NMDAR test results were positive in the CSF (1:40 titer) but negative in the serum. Anti-voltage-gated potassium channel (VGKC), anti-glutamic acid decarboxylase (GAD), antiperoxidase, antithyroglobulin, and paraneoplastic antibodies tested negative in the CSF and serum.

Pelvic computed tomography scans of the axial (A) and coronal (B) views showing a cystic pelvic mass (arrows) with complex components measuring 3.8 cm×4 cm×3.6 cm along the right wall that contains soft tissue fat and calcium, findings that are most consistent with an ovarian dermoid cyst or teratoma.
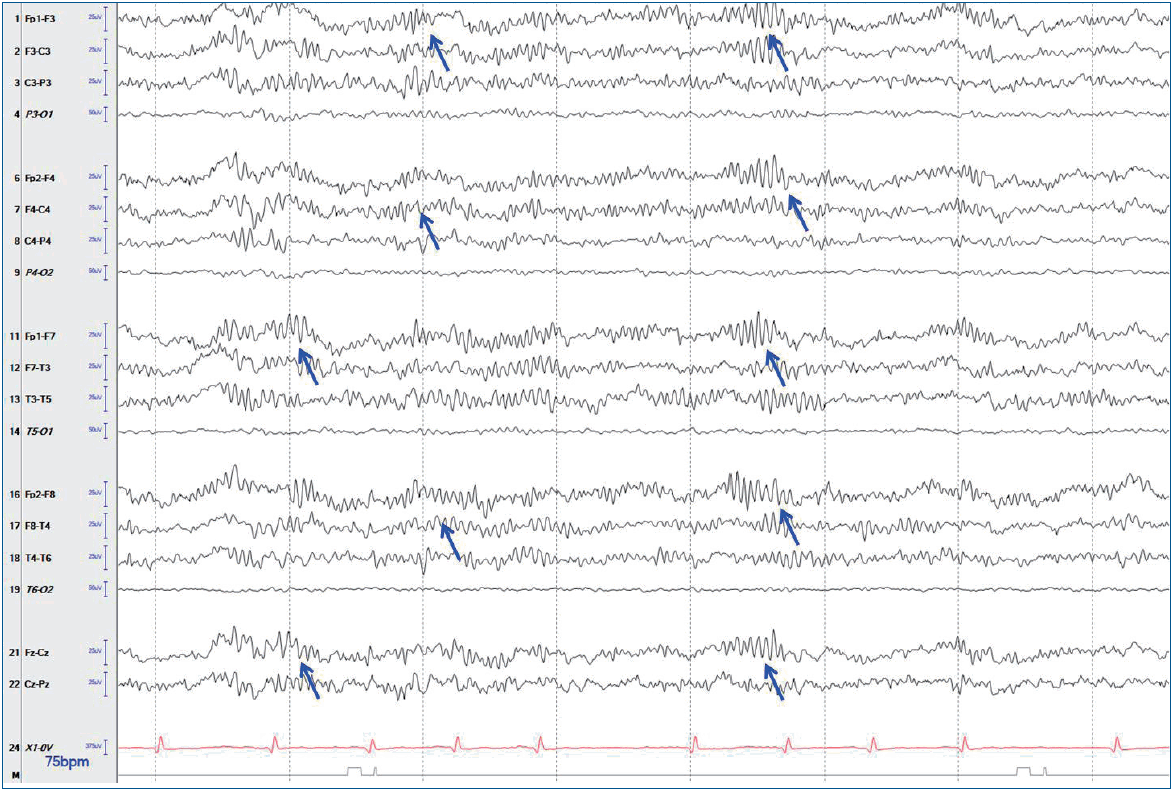
Electroencephalogram (EEG) performed at 5 days after the initial presentation revealing diffuse delta activity with superimposed diffuse rhythmic beta frequency activity (“extreme delta brush” pattern, arrows). The EEG was acquired using the international 10–20 system of electrode placement with longitudinal bipolar montage, which was designed for 18 channels: Fp1-F3, F3-C3, C3-P3, P3-O1, Fp2-F4, F4-C4, C4-P4, P4-O2, Fp1-F7, F7-T3, T3-T5, T5-O1, Fp2-F8, F8-T4, T4-T6, T6-O2, Fz-Cz, and Cz-Pz.
The patient was administered high-dose intravenous (IV) methylprednisolone 20 mg/kg daily for 5 days and IV immunoglobulin G (IVIG) 400 mg/kg daily for 5 days. Her aggressive activity, ataxic gait, and dysarthria symptoms continued to wax and wane since the first-line immune therapy. No significant improvement was observed on video/EEG monitoring. She was treated with IV rituximab (a monoclonal anti-CD20 antibody, 375 mg/m2/wk) as the second-line immune therapy. Her neurological symptoms and behavioral abnormalities improved significantly after IV rituximab therapy, while repeat MRI showed negative results; repeat CSF examination showed decreased anti-NMDAR antibody titer (1:20). Video/EEG findings became normal. She had no significant complications such as a serious infection or viral reactivation during the rituximab treatment. Final pathology of the ovarian mass showed a right cystic teratoma. She was discharged on day 19 with significant improvement in neurologic status and planned to continue active rehabilitation treatment. Presently, she is no longer taking antihypertensive drugs. At the 12-month follow-up, she reported continued improvement in her neurologic symptoms and a stable mood.
Neuron-specific autoantibodies
As with other autoimmune disorders affecting the nervous system, 3 types of autoantibodies have been recognized according to antigen location: (1) those targeting intracellular proteins; (2) those targeting plasma membrane proteins including synaptic receptors; and (3) those targeting ion channels and/or other cell surface proteins (Table 1) [5-7].

Autoimmune or paraneoplastic encephalitis with antibodies against neuron-specific antigens
Antibodies against intracellular antigens are commonly detected in patients with cancer, mainly small-cell carcinoma, and are typical in adults; they are defined as paraneoplastic antibodies but are unlikely to cause neurological manifestations alone [8]. The most important antibodies included in this group are against Ma2, Hu (also defined as type 1 antineuronal nuclear autoantibody), and GAD [6]. The intracellular antigens can be exposed to autoantibodies within the synaptic cleft [9], a cytotoxic T-cell-mediated immune response against neurons, is more likely to be the main mechanism leading to specific damage. In all cases, the response to immunosuppressive therapy is poor.
The pathogenesis of neuronal damage and patterns of response to therapy in autoimmune encephalitis, in which the immune response targets proteins that are part of synaptic receptors, ion channels, or cell surface, is different. Unlike patients with antibodies to intracellular targets, those with antibodies against plasma membrane protein or ion channels respond remarkably well to immunotherapy. These antibodies, disrupting the target antigen, are the true cause of damage [10]. The same phenomenon may occur when drugs or genetic mutations cause disruption of the target antigen. Synaptic receptor antigens involved in autoimmune encephalitis include NMDA, γ-aminobutyric acids A and B receptors (GABAA R, GABAB R), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), metabotropic glutamate receptor 5, and the dopamine 2 receptor [2,6,10]. Antigens that are part of ion channels or the cell surface include leucine-rich glioma inactivated-1 (LGI1), contactin-associated protein-like 2 (Caspr2), dipeptidyl-peptidase-like protein 6 (DPPX), myelin oligodendrocyte glycoprotein, aquaporin 4, and ganglioside GQ1b [2,6,10].
However, identifying antibodies involved in different autoimmune encephalitis is difficult, as it requires complicated laboratory methods that are not readily available in many institutions. Therefore, an autoimmune encephalitis diagnosis should be suspected following a combination of proper signs and symptoms. The initial assessment should include a neurological evaluation and standard diagnostic tests including MRI and CSF study. The diagnosis of autoimmune encephalitis should be considered at all ages when all 3 of the following criteria have been met (Table 2): (1) subacute onset of working memory deficits (short-term memory loss), altered mental status (decreased or altered level of consciousness, lethargy, or personality change), or psychiatric symptoms; (2) at least one of the following: new focal CNS findings; seizures not explained by a previously known seizure disorder; CSF pleocytosis with a WBC count >5/mm3; MRI features suggestive of encephalitis (hyperintense signal highly restricted to one or both medial temporal lobes (as in limbic encephalitis [LE]), or in multifocal areas involving grey matter, white matter, or both compatible with demyelination or inflammation); and (3) reasonable exclusion of alternative causes [6].

Specific autoimmune encephalitis syndrome
Although clinical manifestations of autoimmune encephalitis can vary significantly, different autoantibodies can be associated with similar phenotypes, certain clinical features suggest a specific type of autoimmune encephalitis, such as facio-brachial dystonic seizures (FBDS) and memory impairment with anti-LGI1 encephalitis; encephalopathy, insomnia, ataxia, peripheral nerve hyperexcitability, and neuropathic pain with anti-Caspr2 encephalitis; refractory SE with anti-GABAA R encephalitis; and myoclonus, tremors, hyperexcitability, and severe diarrhea with anti-DPPX encephalitis (Table 1). Seizures and movement disorders along with psychosis, confusion, or additional behavioral changes are the most relevant initial manifestations in all age groups.
1. Anti-NMDAR encephalitis
Anti-NMDAR encephalitis is the most common and best characterized autoimmune encephalitis syndrome [11,12]. Although this encephalitis type can affect people of any age, it is predominant in young women and children. In many patients, both neurological signs and symptoms are associated with those of a tumor, most commonly an ovarian teratoma, although testicular immature teratoma and lymphoma have been also described [11-13].
NMDARs are ligand-gated ion channels that mediate excitatory neurotransmission, are included in a group of ionotropic glutamate receptors with tetrameric complexes of large transmembrane subunits [14]. Three different NMDAR subunits have been identified: NR1, NR2, and NR3. The simultaneous binding of glycine and glutamate activates NMDAR, which results in cation (mainly Ca2+) influx into the cell provided that sufficiently strong depolarization is present. NMDAR signaling is critical for activity-dependent synaptic plasticity and long-term hippocampal potentiation [15]. Moreover, NMDARs modulate excitotoxicity, as excessive cation influx can cause neuronal death [16]. In anti-NMDAR encephalitis, the autoimmune process causes a selective and reversible decrease in NMDAR surface density and synaptic localization, leading to disruption of the synaptic structure and function, strictly dependent on the patient’s antibody titer [17]. An autoimmune response to the NR1 subunit of NMDAR has been shown in children to be triggered by various infectious agents, and different reports have suggested that anti-NMDAR encephalitis may be associated with HSV, Mycoplasma pneumoniae, measles virus, mumps, influenza A/H1N1 infection, group A hemolytic Streptococcus, and Toxoplasma gondii infection [18,19].
In a multicenter observational study of 577 patients, the disease predominantly affected young people with a female sex predominance of 4:1. This predominance was less evident in children aged <12 years and adults aged >45 years [11]. Many patients present with prodromal headache, fever, or a viral-like process, followed in a few days by multistage progression of characteristic symptoms: prominent psychiatric manifestations (anxiety, agitation, bizarre behavior, hallucinations, delusions, disorganized thinking), insomnia, memory deficits, seizures, cognitive impairment, movement disorders (dyskinesias, choreoathetoid, dystonia, tremor, ataxia, rigidity, opisthotonic postures), language dysfunction, autonomic instability (hyperthermia, blood pressure fluctuations, tachycardia, bradycardia, and sometimes hypoventilation requiring mechanical ventilation) [20]. Clinical manifestations differ between adults and children. First, the association with a tumor is less common in children. Approximately 50% of females aged >18 years have uni- or bilateral ovarian teratomas versus <9% of girls aged <14 years [11]. In teenagers and adults, neuropsychiatric manifestations (psychosis, delusion, agitation, aggression, or catatonia), memory loss, and hallucinations are the rule at onset. Contrastingly, only 60% of children present exclusively with psychiatric symptoms, whereas movement disorders and seizures are significantly more common. Seizures can occur at any time but tend to strike earlier in males, being general or focal [21]. Regardless of patient age and presentation, the clinical features at 3–4 weeks after symptom onset are similar in most cases within several categories.
CSF pleocytosis is significantly less common in children than in adults (43% vs. 63%, respectively; P=0.0163) [20]. Almost all patients have abnormal EEG findings at disease onset and peak. Diffuse slowing is the most common feature in afflicted children and adults [22]. Moreover, at the peak stage, focal slowing and epileptiform discharges, polymorphic delta rhythm, diffuse fast activities, and extreme delta brush can also be detected [23]. MRI findings are often normal or show medial temporal and frontal hyperintensity on T2-weighted images and leptomeningeal contrast enhancement [24]. Subcortical white matter changes seem as common as those of cortical grey matter. Positron emission tomography (PET) can show CNS abnormalities in cases with normal MRI findings [24]. However, very few studies have used this diagnostic tool in patients with anti-NMDAR encephalitis; thus, further research is needed to evaluate the role of PET in diagnosing this syndrome. An anti-NMDAR encephalitis diagnosis is confirmed by the detection of immunoglobulin G (IgG) antibodies to the GluN1 (also known as NR1) subunit of NMDAR in the serum or CSF [25]. CSF IgG antibody testing is highly sensitive and specific for anti-NMDAR encephalitis; false-positive and -negative results may occur when only serum is tested. While waiting for confirmatory IgG anti-Glu1 antibody results, we should assess a patient with promptly progressive encephalopathy as having probable anti-NMDAR encephalitis if they fulfill the criteria shown in Table 3 [6].
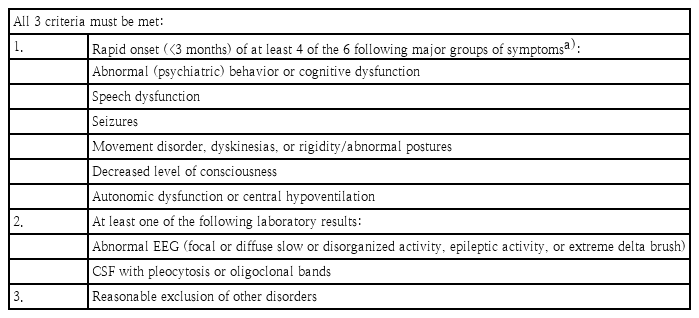
Diagnostic criteria for probable anti-NMDA receptor encephalitis
Treatment options include immunosuppression and tumor resection when indicated [11,26]. In the absence of prospective and randomized data, treatment decisions should be individualized and consider patient age, tumor presence or absence, and symptom severity. Based on the observational studies reviewed below and our clinical experience, we recommend first-line immunotherapy involving IV methylprednisolone (e.g., 1 g daily for 5 days in an adult), IVIG (e.g., 400 mg/kg/day for 5 days) or plasma exchange alone or combined with tumor removal, when appropriate. It is unknown whether IVIG and plasma exchange have similar efficacies. If there is no evidence of clinical improvement with initial therapies, we progress to second-line therapies including rituximab (either 375 mg/m2 weekly for 4 weeks, or 1 g twice 2 weeks apart), and cyclophosphamide (750 mg/m2 monthly for 4–6 months depending on the results) alone or combined. These second-line drugs are recommended if no sustained improvement occurs within 4 weeks of immunotherapy initiation or tumor removal [11]. Factors associated with better prognosis included no need for intensive care unit admission (P<0.0001), early treatment (P=0.009), and low disease severity within 4 weeks from onset (P=0.011) [5].
2. VGKC-positive group of autoimmune encephalitis
These diseases start as LE with cognitive impairment, seizures, and variable neuropsychiatric symptoms.
1) Anti-LGI1 encephalitis
The associated antibodies target the secreted neuronal protein LGI1, functioning as a ligand for 2 epilepsy-related proteins, ADAM22 and ADAM23, that are essential for inhibitory signal transmission from the presynaptic potassium channel to the postsynaptic AMPA receptors [27,28]. Antibody binding to LGI1 disrupts pre- and postsynaptic LGI1 signaling, resulting in neuronal hyperexcitability [29].
Anti-LGI1 encephalitis occurs in approximately 60-year-old men presenting with LE. The clinical features are homogeneous and include seizures and subacute progressive memory alterations; behavior and space orientation abnormalities are the most common signs. In some cases, seizures precede the cognitive decline [30]. The most characteristic seizures, detectable in approximately 50% of cases, are FBDS. FBDS, considered pathognomonic of anti-LGI1 encephalitis, are involuntary unilateral contractions of the face and arm (or leg) that last for <3 seconds and occur up to 100 times a day. Generalized tonic-clonic seizures can also be seen in the late disease stage. Memory and cognitive deficits may be preceded by short FBDS [31,32]. Patients can develop hyponatremia (nearly 60%) and rapid eye movement sleep behavior disorder. EEG can reveal the multifocal interictal epileptiform discharges and slow waves. MRI usually reveals findings typical of LE (e.g., medial temporal lobe hyperintensity), while CSF findings are often normal or include only oligoclonal bands[33,34]. Approximately 5%–11% of cases are associated with cancer; the most common associated tumor is thymoma. The association with other tumors may be coincidental [30].
Treatment with glucocorticoids, IVIG, mycophenolate mofetil, and/or plasma exchange results in significant improvement in 70%–80% of patients [31]. The early immunotherapy initiation in patients with FBDS may prevent cognitive impairment and improve long-term outcomes [32]. However, experience with rituximab as an add-on therapy is limited [35]. Relapses occur in up to one-third of patients and are associated with worse outcomes. Despite substantial recovery, cognitive deficits and disability persist in many patients along with evidence of hippocampal atrophy on MRI [36].
2) Anti-Caspr2-associated encephalitis
The target antigen is Caspr2, an adhesion molecule expressed in the CNS and peripheral nervous system (PNS) that is essential for maintaining normal function of the VGKC [37]. Anti-Caspr2-associated encephalitis can manifest as LE: Morvan syndrome (neuromyotonia, memory loss and confusion, sleep disturbances, autonomic dysfunction), and in few patients, as isolated neuromyotonia [34,37,38].
This disorder predominantly affects older men (median age, 65 years), although rare pediatric cases have been described [38,39].) The presentation and disease course are slower than those of other autoimmune encephalitis syndromes. Although the clinical syndrome is less homogeneous than anti-LGI1 encephalitis, almost 80% of patients develop ≥3 of the core symptoms (“strongly supported diagnostic symptoms”): encephalopathy (cognitive decline/seizures), cerebellar symptoms, PNS hyperexcitability, autonomic dysfunction, insomnia, neuropathic pain, and weight loss, symptoms that might progress very slowly, mimicking dementia [39,40].) Due to PNS involvement, muscular pain, fasciculations, and cramps are present in many patients. The disorder is usually not associated with cancer. A tumor, usually thymoma, may be revealed in up to 32% of cases [41]. Patients with tumors (commonly thymoma) are more likely to develop Morvan syndrome than those with isolated central or peripheral symptoms. CSF pleocytosis is seen in 30%–40% of cases. MRI findings are normal in most patients or consist of bilateral T2 hyperintensity of the medial temporal lobes.
Tumor removal is crucial for improvement and cure. Immunotherapy seems effective in patients without malignancy. However, relapse affects approximately 25%, occurring even several years after the initial episode and involving other areas of the CNS [39]. The presence of autoantibodies directed against the synaptic protein LGI1 and adhesion molecule Caspr2 has not been found in children with acute encephalitis; thus, the role of these autoantibodies in the pediatric population is still debated.
3) VGKC-positive encephalitis in the absence of anti-LGI1 and Caspr2 antibodies.
The clinical features of antibodies against VGKC in patients lacking anti-LGI1 and anti-Caspr2 antibodies has not been precisely defined. These patients have a heterogeneous clinical spectrum, including pain syndrome, LE, epilepsy, polyneuropathy, and cramp fasciculation syndromes, all associated with the demonstration of anti-VGKC autoantibodies [42]. An acute and isolated psychotic manifestation of anti-VGKC encephalitis was recently reported in a 16-year-old girl, but further studies are required to recognize potential new antibodies against neuronal structures [43].
3. Anti-GABAR encephalitis
Neurotransmitter γ-aminobutyric acid (GABA) is the chief inhibitory neurotransmitter in the vertebrate CNS. GABAA R are ligand-gated ion channels that allow the influx of Cl− and fast synaptic inhibition; metabotropic GABAB R are expressed in the CNS and PNS: This effect is primarily inhibitory via the inhibition of presynaptic voltage-gated Ca2+ channels, activation of postsynaptic K+ channels, and inhibition of adenylyl cyclase [44].
Patients with anti-GABAA R encephalitis develop rapidly progressive encephalitis with refractory seizures, SE, and/or epilepsia partialis continua [45]. Nearly 50% of reported cases involve children [46]. CSF often shows lymphocytic pleocytosis with increased protein concentrations. Unlike other causes of autoimmune encephalitis, the MRI in anti-GABAA R encephalitis often shows multifocal cortical/subcortical and widespread abnormal signaling on fluid-attenuated inversion recovery (FLAIR) and T2-weighted scans [46]. Tumors (usually thymoma) occur in 40% of patients, almost all of whom are adults [45,46]. In children, anti-GABAA R encephalitis may progress as a postviral encephalitis and coexist with NMDAR antibodies. A case of a previously healthy child presenting with catatonia and encephalopathy without seizures due to antibodies against GABAA R was recently reported [47]. Patients respond to immunotherapy but often require pharmacologic-induced coma for prolonged seizures.
Encephalitis due to antibodies against the GABAB R was described primarily in adults presenting with LE, including seiures, SE, ataxia, or opsoclonus-myoclonus [48]. One pediatric case was characterized by encephalopathy, refractory seizures, and a mixed movement disorder (opsoclonus, ataxia, and chorea) [49]. Approximately 50% of cases are paraneoplastic and almost always associated with small-cell lung cancer [48]. MRI reveals FLAIR and T2 hyperintensity consistent with LE in approximately 50%; over one-half of patients have CSF pleocytosis and/or elevated protein levels [48]. The response to immunosuppressive therapies was good in tumor-free patients.
4. Anti-AMPA receptor encephalitis
Encephalitis associated with antibodies against the AMPA receptor predominantly affects females, with a median age of onset of 50–60 years [50,51]. In a case series of 22 patients with anti-AMPA receptor antibodies, LE with or without seizures was the most common clinical presentation (55%); other manifestations included limbic dysfunction along with diffuse encephalopathy (36%), motor deficits followed by LE (one patient), and psychosis with bipolar features (one patient) [51]. An underlying tumor is detected in approximately two-thirds of the patients, most commonly in the lung, thymus, or breast [50,51]. CSF lymphocytic pleocytosis has been described in 50% to 90% (WBC count, 5–164/mm3) [50,51]. MRI findings are usually abnormal, with FLAIR signal abnormalities in the medial temporal lobes. In one study, 9 of 10 patients responded to the tumor resection and/or immunotherapy, but consequent relapses occurred in 5 (in the absence of tumor recurrence) and were associated with an incomplete treatment response and death of SE in one patient [50].
5. Anti-glycine receptor encephalopathy
Glycine receptors (GlyRs) are ligand-gated chloride channels that mediate fast inhibitory transmission, leading to neural hyperpolarization through a selective influx of chloride. GlyRs are essential for muscle tone, coordination, respiratory rhythm, and sensory processing [52]. The genetic modification of GlyRs is associated with hypertonia, hyperekplexia, development delay, apnea, and stiff-person syndrome in some patients [53]. Patients with anti-GlyR antibodies often have progressive encephalomyelitis with rigidity and myoclonus, although severity and clinical expressions vary [54]. A few patients have been reported with limbic or other encephalopathy without brainstem or spinal cord features. In one study of 45 patients, 5 had a past history and successful treatment of a tumor, while 4 were concurrently diagnosed with tumor and neurologic disease [55]. They reported that, at onset, spasms, which were often painful, and stiffness/rigidity of the neck, trunk, or limb muscles were demonstrated in 69% of patients, whereas diplopia, ptosis, and nystagmus were found in 40% of patients. Finally, cognitive impairment was observed in 29%, while seizures occurred in 13%.
6. Anti-GAD encephalitis
GAD is an intracellular enzyme that is found in neural and pancreatic cells [56]. Two isoforms of GAD (GAD 65 and GAD 67) have been identified; both can be detected in the CNS, where they catalyze GABA (the major inhibitory neurotransmitter) synthesis, whereas pancreatic islet cells contain only GAD [65.56]. Antibodies against GAD are associated with both type 1 diabetes and several neurological manifestations, including LE, stiff-person syndrome, cerebellar ataxia, ocular movement disorders, and refractory epilepsy [56]. Very few cases have been found in children. In particular, LE with anti-GAD 65 antibodies is characterized by rapidly progressive short-term memory loss, psychiatric symptoms, and seizures; it commonly has a chronic course with poor response to immunotherapy and AEDs [57,58]. Stiff-person syndrome with abnormal postures (often hunched over and stiffened) are characteristics of this disorder. Malignancy (thymoma or small-cell lung carcinoma) is comorbid with approximately 25% of cases of anti-GAD 65 encephalitis [6]. Recently, a 9-year-old girl was reported with refractory seizures and behavioral and severe fatal autonomic dysfunction, including arrhythmia, bradycardia/tachycardia, hypotension/hypertension, hypothermia/hyperthermia, and hyperhidrosis, demonstrating the possibility of extralimbic brain involvement in children with anti-GAD 65 antibodies [59].
Diagnostic approach
In the appropriate clinical presentation, detecting specific autoantibodies confirms the autoimmune encephalitis diagnosis. However, not all patients with paraneoplastic or autoimmune encephalitis have antibodies, and the absence of antibodies does not rule out an autoimmune mechanism. For patients without antibodies, the suspicion of an autoimmune or paraneoplastic etiology is based on the clinical history of a subacute progressive neurologic course, cancer risk factors, and supportive laboratory and radiologic results.
The differential diagnosis of autoimmune or paraneoplastic encephalitis includes various broad categories, such as infection, toxic and metabolic disturbances, vascular disorders, neoplastic disorders, demyelinating and inflammatory disorders, psychiatric diseases, neurodegenerative dementias, and rare heritable or metabolic disorders (Table 4) [25,60]. Patients with suspected autoimmune encephalitis (Table 2) should undergo neuroimaging, EEG, lumbar puncture, and serologic testing for appropriate biomarkers to confirm the diagnosis and exclude alternative etiologies.

Differential diagnosis of autoimmune encephalitis
Distinctive MRI findings of autoimmune encephalitis include signal hyperintensities on FLAIR or T2-weighted images in affected brain regions (e.g., medial temporal lobes and/or brainstem, subcortical regions, and cerebellum). Contrast enhancement is variable [45]. EEG should be performed to exclude nonconvulsive seizures. Nonspecific abnormal findings are common and include focal or generalized slowing, epileptiform discharges, and periodic lateralized epileptiform discharges [61]. Approximately one-third of patients with NMDAR encephalitis have the finding of extreme delta brush, considered a characteristic of the disorder [23]. The mechanism underlying the delta brush remains unclear. As a cellular mechanism, the modulation of NMDAR-mediated currents was proposed by altering rhythmic neuronal activity and leading to this pattern [23]. Furthermore, delta brush was recently found among intracranial EEG ictal-onset patterns related to focal cortical dysplasia in a series including hippocampal sclerosis with different epileptogenic lesions. Therefore, delta brush is not pathognomonic for NMDAR encephalitis. But its presence still can guide early recognition and prompt immunotherapy in NMDAR encephalitis, especially in patients with proper clinical features when other possible etiologies are excluded [62]. A CSF examination should be performed to exclude viral infection and leptomeningeal metastasis. Patients with autoimmune encephalitis may or may not have abnormal CSF findings. Abnormalities include modestly elevated protein (<100 mg/dL), mild to moderate lymphocytic pleocytosis, an elevated IgG index, and/or the presence of oligoclonal bands [61,63]. However, these findings are variable. The absence of inflammatory evidence on CSF and MRI may be common in older adults [64]. Screening for malignancy is mandatory in children to rule out a neoplastic syndrome.
Therapeutic approach
Immunosuppressive therapy should not be delayed until a cancer diagnosis confirmation or antibody results in patients with a typical paraneoplastic syndrome or autoimmune encephalitis provided that an underlying infectious etiology has been ruled out and there are no other contraindications [65]. There are no evidence-based treatment standards for autoimmune encephalitis, although many clinicians have become confident with the prompt use of high-dose methylprednisolone combined with IVIG [66]. Plasma exchange is also useful first-line acute treatment for critically ill patients or when IV steroid or IVIG is poorly tolerated. After an initial trial of therapy, patients should be reevaluated for clinical improvement. Rituximab or cyclophosphamide can be considered second-line agents when there is no or an incomplete response to first-line treatment. SE in a patient with anti-NMDAR antibodies or LE and LGI1 positivity may more rapidly escalate immunotherapy.
Other immunotherapies could be considered in post-infectious cases such as immunoadsorption (antibody-removing therapy) alternative to plasma exchange, which should be started as promptly as possible to warrant the best neurological outcomes, although several weeks or months might be required to show clear effectiveness [67]. More research on differential blood-brain barrier permeability would resolve many questions about autoantibody entry and the pathogenesis of autoimmune encephalitis. There are no data about anticytokine therapies in young patients with refractory autoimmune encephalitis. Overall outcomes are usually good in younger patients with autoimmune encephalitis, although this depends on the underlying tumor and stage as well as the severity of the initial neurological symptoms [68]. The delay to diagnosis and treatment has been associated with worse prognosis and increased relapse rates [11].
Conclusion
The proper diagnosis and prompt management of autoimmune encephalitis require an organized approach. The evaluation should begin with a detailed history and physical examination to detect clues to specific causes. Ancillary MRI, EEG, and CSF testing may further support a diagnosis of encephalitis and exclude infectious causes. Antineuronal antibody tests are used to make the diagnosis, but a negative antibody test may not exclude autoimmune disorders indefinitely. Treatment for suspected autoimmune encephalitis could be considered prior to antibody test results are available since prompt treatment is associated with better outcomes.
Notes
No potential conflict of interest relevant to this article was reported.