Changes of Bax, Bcl-2, CCR-2, MCP-1, and TGF-β1 genes in the left ventricle of spontaneously hypertensive rat after losartan treatment
Article information

Abstract
Purpose
Increased apoptosis was recently found in the hypertrophied left ventricle of spontaneously hypertensive rats (SHRs). Although the available evidence suggests that apoptosis can be induced in cardiac cells by various insults including pressure overload, cardiac apoptosis appears to result from an exaggerated local production of angiotensin in adult SHRs. Altered expressions of Bcl associated X (Bax), Bcl-2, chemokine receptor (CCR)-2, monocyte chemoattractant protein (MCP)-1, transforming growth factor (TGF)-β1, phosphorylated extracellular signal-regulated kinases (PERK), and connexin 43 proteins, and kallikrein mRNA were investigated to explore the effects of losartan on the SHR model.
Methods
Twelve-week-old male rats were grouped as follows: control (C), SHR (hypertension: H), and losartan (L; SHRs were treated with losartan [10 mg/kg/day] for 5 weeks). Western blot and reverse transcription polymerase chain reaction assays were performed.
Results
Expression of Bax, CCR-2, MCP-1, TGF-β1, PERK, and connexin 43 proteins, and kallikrein mRNA was significantly increased in the H group compared to that in the C group at weeks 3 and 5. Expression of Bax, CCR-2, MCP-1, TGF-β1, and connexin 43 proteins and kallikrein mRNA was significantly decreased after losartan treatment at week 5. PERK protein expression was significantly decreased after losartan treatment at weeks 3 and 5. Bcl-2 protein expression was significantly decreased in the H group compared to that in the C group at weeks 3 and 5.
Conclusion
Losartan treatment reduced expression of Bax, CCR-2, MCP-1, TGF-β1, PERK, and connexin 43 proteins, and kallikrein mRNA in SHRs, along with decreased inflammation and apoptosis.
Introduction
Hypertension continues to be a significant clinical problem that can affect quality of life. When uncontrolled, it may contribute to the occurrence of other diseases, such as atherosclerosis [1]. Apoptosis and inflammation contribute to cardiac hypertrophy in hypertension [2]. Alteration in apoptosis has been noted in a variety of tissues in spontaneously hypertensive rats (SHRs) and angiotensin-infused rat models [3]. There are a number of apoptosis modulators and apoptosis modulator candidates, including angiotensin II receptors [4].
Inflammation plays an important role in in triggering fibrosis and cardiac remodeling [5,6]. Sustained systemic arterial hypertension causes progressive myocardial remodeling. Left ventricular (LV) remodeling is characterized by the combination of cardiomyocyte hypertrophy and proliferation of tissues in the LV of SHR [7]. Increased proinflammatory markers such as interleukin-6 and tumor necrosis factor-α have been found in the LV of SHR [8]. However, the molecular mechanism by which hypertension leads to apoptosis and inflammation is not well understood.
Apoptosis, inflammation and fibrosis contribute to vascular remodeling in hypertension [9]. Monocyte chemoattractant protein (MCP)-1 is an inflammatory cytokine, and chemoattractant receptor (CCR)-2 is one of its receptors [9]. Tumor growth factor (TGF)-β is a locally generated cytokine that contributes to tissue fibrosis in various organ systems. Increased cardiac TGF-β levels may be the result of increased cardiac radical oxygen species formation, thus promoting fibrosis during all phases of hypertension [10].
The endoplasmic reticulum (ER) plays an essential role in controlling various intracellular physiological functions by stimulating the signaling networks that control either cell survival or death. Prolonged ER stress induces oxidative stress, inflammation, and apoptotic response. It is highly associated with cardiovascular diseases such as cardiac hypertrophy, heart failure, atherosclerosis, and ischemic heart disease [11]. In particular, ER stress-induced endothelial cell damage and dysfunction exert negative influences on cardiovascular diseases through apoptosis and inflammation [12].
Connexin 43 is a protein involved in gap junction and many physiological processes including smooth muscle depolarization [13]. Deficiency of connexin 43 is related to pathological alterations in SHR. Connexin 43 double knockout mice showed symptoms of obstructed right ventricular outflow tract, cardiac malformation and neonatal death [13].
The kallikrein-kinin and renin-angiotensin systems play a key role in multiple physiological and pathophysiological conditions, including blood pressure regulation, vascular smooth muscle cells growth, and inflammation [14].
SHR is considered a reliable model because the weight of LV rapidly increases in response to the progression of the disease [15]. In addition, the effects of various antihypertensive drugs have been evaluated through this model [16].
Angiotensin II plays an important role in hypertension as it induces changes in inflammation, apoptosis, smooth muscle cell growth and the deposition of extracellular matrix. Angiotensin II is a vasoactive peptide which has a variety of effects in hypertension [17,18]. Angiotensin II induces inflammation through the inflammatory cytokines such as MCP-1 [19]. In response to angiotensin II, monocytes increase MCP-1 secretion. In addition, CCR-2, a receptor of MCP-1, is involved in angiotensin II-induced atherosclerosis [20].
Losartan is a selective angiotensin II receptor antagonist [21]. Losartan is used clinically for reducing arterial blood pressure; it also has an important role in apoptosis and inflammation. We confirmed attenuated LV hypertrophy by 10-mg/kg/day losartan administration in SHR in a previous study [22]. However, underlying molecular mechanism is not entirely clear.
The purpose of this study is to investigate the effect of losartan on cardiac apoptosis, inflammation, function, kallikrein-kinin system related protein, and mRNA expression levels in the LV of SHR models.
Materials and methods
1. Animal experiment
Twelve-week-old male SHR and normotensive Wistar-Kyoto rats were used for this research. The rats were assigned to 3 experimental groups with within-cage randomization. Groups were divided as follows: control (C, n=9) group, SHR (H, n=15) group, and losartan (L, n=18) group. L group was fed losartan 10 mg/kg/day by gavage feeding for 5 weeks. All rats were housed in a temperature-controlled environment with free access to rodent chow and autoclaved water.
Before they were sacrificed, the rats were anesthetized with zoletil (Virbac, Carros, France) and rompun (Bayer Korea, Seoul, Korea) intraperitoneally. LVs were separated from right ventricles and quickly frozen in liquid nitrogen for RNA and protein extraction.
All the experimental procedures were approved by the Institutional Animal Care and Use Committee of Ewha Womans University School of Medicine (approval number: 13-0125).
2. Western blot analysis
LV homogenates were subjected to protein analysis. The isolated LV was lysed in 4˚C Proprep (iNtRON Biotechnology Inc., Seongnam, Korea) for 1 hour and centrifuged at 12,000 rpm in 4˚C for 30 minutes. Protein concentration in supernatant was measured by bicinchoninic acid assay. Thirty microgram of each sample was boiled for 10 minutes and loaded onto 10% sodium dodecyl sulfate polyacrylamide gel. It was electrically transferred onto nitrocellulose membrane, and the membranes were incubated in a 5% bovine serum albumin in tris-buffered saline with Tween 20 (TBST) for 1 hour. Then the membranes were incubated with antibodies specific to Bax, Bcl-2, CCR-2, MCP-1, TGF-β1 and phosphorylated extracellular signal-regulated kinases (PERK) at 4˚C overnight. The membranes were washed 4 times with TBST, and then incubated with secondary antibody for 1 hour. The protein bands were visualized by enhanced chemiluminescence.
3. Polymerase chain reaction
LV homogenates were subjected to gene expression analysis of kallikrein. For this purpose, total RNA extraction, cDNA synthesis, and polymerase chain reactions (PCRs) were performed. RNA extraction was performed with the RNeasy kit (Qiagen, Germantown, MD, USA), and cDNA synthesis was performed using 1 μg of the RNA.
PCR primers for kallikrein were as follows; forward primer: 5’-CCTCCTGGTCAGTCTCGGATT-3’, reverse primer: 5’-TGTCCTGGAGTCATTCCTGTC-3’. PCR was performed using G-taq PCR premix kit (Cosmo-genetech Co., Ltd., Seoul, Korea) under the following conditions: initial denaturation at 95˚C for 5 minutes, then 30 cycles of 94˚C for 30 seconds, 59˚C for 30 seconds, and 72˚C for 30 seconds. The amplified PCR products were separated on 1% agarose gels.
4. Statistical analysis
Animals were randomized within each experimental group. Results were expressed as the mean±standard deviation. A Kruskal-Wallis test was used for the comparison of differences in the 3 groups and a Mann-Whitney test was used for between groups comparisons with Bonferroni correction. P value of <0.05 was considered statistically significant. SPSS ver. 14.0 (SPSS Inc., Chicago, IL, USA) was used for all statistical analyses.
Results
1. Losartan decreased apoptosis in LV tissues of SHR
To investigate apoptosis in LV tissues, we confirmed protein expression levels of Bax and Bcl-2. Bax was significantly increased in the H group compared to the C group at weeks 3 (C vs. H group: 0.19±0.02 vs. 0.88±0.02, P<0.05) and 5 (C vs. H group: 0.20±0.02 vs. 0.92±0.04, P<0.05). Losartan significantly decreased the expression levels of Bax at week 5 (H vs. L group: 0.92±0.04 vs. 0.84±0.05, P<0.05) (Fig. 1A). Bcl-2 was significantly decreased in the H group compared to the C group at weeks 3 (C vs. H group: 0.87±0.03 vs. 0.23±0.02, P<0.05) and 5 (C vs. H group: 0.80±0.03 vs. 0.51±0.05, P<0.05). There was no significant change after losartan treatment (Fig. 1B). Apoptosis was significantly decreased after losartan treatment.
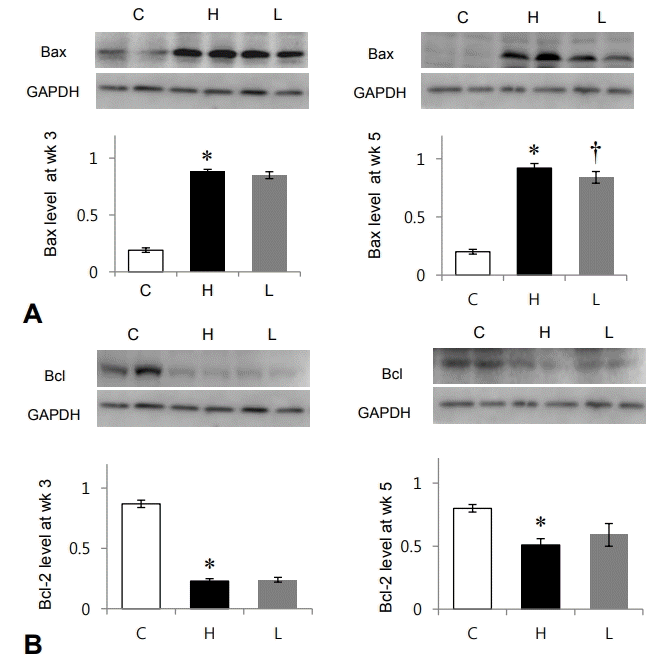
Changes in apoptosis-related protein expression after losartan treatment in spontaneously hypertensive rats. Protein expression level of Bax was significantly decreased in the L group compared to that in the H group at week 5. Protein expression levels of Bcl-2 showed no significant changes between the H and L groups. Bax, Bcl associated X; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; C, control group; H, spontaneously hypertensive rat group; L, losartan group. *Significantly different between C and M groups, P<0.05. †Significantly different between H and L groups, P<0.05.
2. Losartan decreased inflammation in the LV tissues of SHR
Protein expression level of CCR-2 was significantly increased in the H group compared to the C group at weeks 3 (C vs. H group: 0.17±0.01 vs. 0.86±0.02, P<0.05) and 5 (C vs. H group: 0.17±0.01 vs. 0.91±0.05, P<0.05). CCR-2 was significantly decreased at week 5 after losartan treatment (H vs. L group: 0.91±0.05 vs. 0.79±0.04, P<0.05) (Fig. 2A).

Changes in inflammation-related protein expression after losartan treatment in spontaneously hypertensive rats. Protein expression levels of CCR-2, MCP-1, and TGF-β1 were significantly decreased in the L group compared to those in the H group at week 5. CCR-2, chemokine receptor-2; MCP-1, monocyte chemoattractant protein-1; TGF-β1, transforming growth factor-beta 1; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; C, control group; H, spontaneously hypertensive rat group; L, losartan group. *Significantly different between C and H groups, P<0.05, †Significantly different between H and L groups, P<0.05.
Protein expression level of MCP-1 was significantly increased in the H group compared to the C group at weeks 3 (C vs. H group: 0.17±0.02 vs. 0.87±0.02, P<0.05) and 5 (C vs. H group: 0.18±0.02 vs. 0.84±0.06, P<0.05) (Fig. 2B). MCP-1 was significantly decreased at week 5 after losartan treatment (H vs. L group: 0.84±0.06 vs. 0.73±0.03, P<0.05) (Fig. 2B).
Protein expression level of TGF-β was significantly increased in the H group compared to the C group at weeks 3 (C vs. H group: 0.17±0.02 vs. 0.86±0.02, P<0.05) and 5 (C vs. H group: 0.19±0.02 vs. 0.77±0.03, P<0.05) (Fig. 2C). TGF-β also decreased 5 weeks after losartan treatment (H vs. L group: 0.77±0.03 vs. 0.65±0.05. P<0.05) (Fig. 2C).
Inflammatory factors, including CCR-2, MCP-1 and TGF-β, were significantly decreased after losartan treatment at week 5.
3. Losartan decreased protein expression levels of PERK in the LV tissues of SHR
Protein expression levels of PERK was significantly increased in the H group compared to the C group at weeks 3 (C vs. H group: 0.14±0.0 vs. 0.65±0.03, P<0.05) and 5 (C vs. H group: 0.28±0.03 vs. 0.88±0.06, P<0.05). PERK was significantly decreased after losartan treatment at weeks 3 (H vs. L group: 0.65±0.03 vs. 0.58±0.03, P<0.05) and 5 (H vs. L group: 0.88±0.06 vs. 0.73±0.05, P<0.05) (Fig. 3).

Changes in phosphorylated extracellular signal-regulated kinases (PERK) protein expression after losartan treatment in spontaneously hypertensive rats. Protein expression level of PERK was significantly decreased in the L group compared to that in the H group at weeks 3 and 5. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; C, control group; H, spontaneously hypertensive rat group; L, losartan group. *Significantly different between C and H groups, P<0.05, †Significantly different between H and L groups, P<0.05.
4. Losartan decreased protein expression levels of connexin 43 in the LV tissues of SHR
Protein expression level of connexin 43 was significantly increased in the H group compared with the C group at weeks 3 (C vs. H group: 0.19±0.02 vs. 0.53±0.03, P<0.05) and 5 (C vs. H group: 0.17±0.01 vs. 0.59±0.0, P<0.05 (Fig. 4). Connexin 43 was reduced in the L group at week 5 (H vs. L group: 0.59±0.04 vs. 0.49±0.04, P<0.05) (Fig. 4).
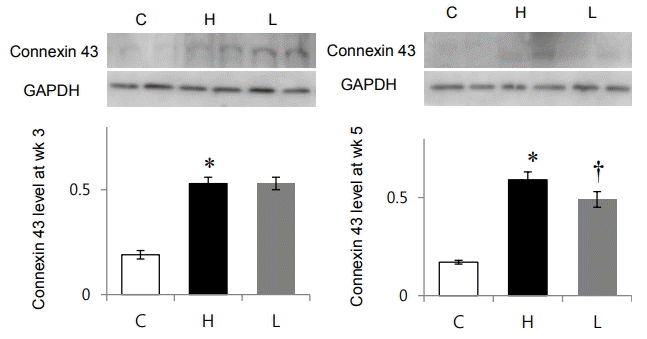
Changes in connexin 43 protein expression after losartan treatment in spontaneously hypertensive rats. Protein expression level of connexin 43 was significantly decreased in the L group compared to that in the H group at week 5. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; C, control group; H, spontaneously hypertensive rat group; L, losartan group. *Significantly different between C and H groups, P<0.05, †Significantly different between H and L groups, P<0.05.
5. Losartan decreased mRNA levels of kallikrein in LV tissues of SHR.
mRNA expression levels of kallikrein was significantly increased in the H group compared to the C group at weeks 3 (C vs. H group: 0.17±0.01 vs. 0.7±0.05, P<0.05) and 5 (C vs. H group: 0.17±0.03 vs. 0.76±0.03, P<0.05) (Fig. 5). Kallikrein mRNA expression levels were significantly decreased in the L group at week 5 (0.76±0.03 vs. 0.61±0.02, P<0.05) (Fig. 5).
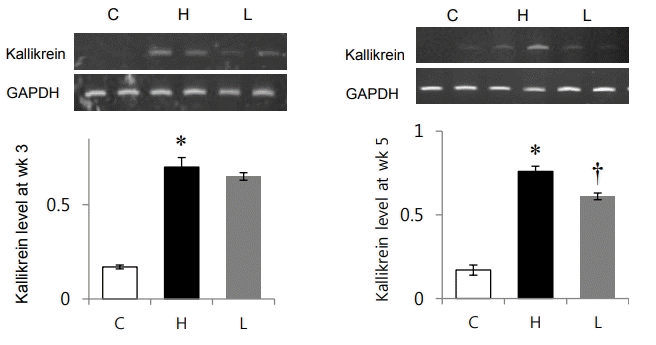
Changes in kallikrein mRNA expression after losartan treatment in spontaneously hypertensive rats. mRNA expression level of kallikrein was significantly decreased in the L group compared to that in the H group at week 5. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; C, control group; H, spontaneously hypertensive rat group; L, losartan group. *Significantly different between C and H groups, P<0.05, †Significantly different between H and L groups, P<0.05.
Discussion
Our main conclusion is that inflammation and apoptosis were increased in the LV of SHR, and losartan reduced these changes.
In our SHR model used in this study, blood pressure was higher than 200 mmHg [22]. In our previous study, we confirmed that LV/body weight ratio was significantly higher in the H group than the C group at weeks 3 and 5 [22]. In this present study, we focus on changes of apoptosis and inflammatory gene expressions after losartan treatment.
Increased Bax protein expression levels and decreased Bcl-2 were observed in the SHR model. It is well known that hypertension is associated with apoptosis, but the role of apoptosis in vascular remodeling has remained unclear. Clarification is needed on whether apoptosis is a growth-related compensatory mechanism or a primary process.
Apoptosis is a physiological, active, and tightly regulated process in which cell death follows a programmed sequence of events. Apoptosis plays a role in the regulation of cell mass and architecture in many tissues. The Bcl-2 is critical for the regulation of apoptosis as it inhibits apoptosis; Bax, on the other hand, induces apoptosis [23].
Increased CCR-2, MCP-1, and TGF-β1 protein expression levels were also noted in the LV of our SHR model. MCP-1 and CCR-2 are important chemokines which regulate the migration and infiltration of monocytes and macrophages. MCP-1 is an inflammatory cytokine and CCR-2 is one of its receptors. Inflammation may play an important role in triggering fibrosis and remodeling in hypertension [9]. CCR-2 is expressed on the monocyte surface. CCR-2 has been found to be induced and involved in a number of diseases including hypertension [24]. Migration of monocytes from the blood stream across the vascular endothelium is required for routine immunological surveillance of tissues, as well as in response to inflammation [25]. An excessive infiltration of monocytes generates more inflammation and increases the risk of heart failure [24].
TGF-β is a locally generated cytokine that contributes to tissue fibrosis in various organ systems. Therefore, the increased cardiac TGF-β levels may be the result of increased cardiac radical oxygen species formation, thus promoting fibrosis during all phases of hypertension [10]. Inflammation, which may be low grade, probably plays an important role in triggering fibrosis in cardiovascular disease and hypertension [9]. Losartan decreased inflammatory protein expression levels such as CCR2, MCP-1, and TGF-β1 in our study. Therefore, losartan is considered to have anti-inflammatory effects on the LV of SHR model.
Increased PERK protein expression levels are also observed in our SHR model. ER stress, which is highly associated with cardiovascular disease, is triggered by a disturbance in ER function because of protein misfolding or an increase in protein secretion. Prolonged disruption of ER causes ER stress and activation of the unfolded protein response, leading to various diseases. ER stress and inflammation signaling pathways are connected through various mechanisms that can induce cardiovascular disease. In atherosclerosis, increased PERK and IRE1/TRAF2 and accumulated ROS activate and augment inflammatory response [26]. Alleviation of ER stress has recently been accepted as a potential therapeutic target to treat cardiovascular diseases such as heart failure, hypertension, and atherosclerosis [11]. Losartan reduced PERK protein level in our study.
We performed this study to further evaluate our previous research result that showed renin-angiotensin system plays an important role in systemic hypertension. Especially, angiotensin II is critical in vasoconstriction, endothelial dysfunction, inflammation and smooth muscle cell growth [27-29]. In addition, angiotensin II binds through its interaction with the angiotensin II type 1 receptor. Therefore, we examined the effects of angiotensin II receptor antagonist, losartan, on SHR and found the following important results.
Losartan has antihypertensive effects. In our previous study, we confirmed that systolic blood pressure was significantly decreased after 3 weeks administration of losartan [30]. Losartan also decreased the protein expression levels of angiotensin II receptor type 1A in the LV of SHR.
These effects of losartan administration on hypertension may be reasonably explained by decreasing apoptosis and inflammation. It is already known that angiotensin II has an important role in SHR. Angiotensin II stimulates smooth muscle apoptosis in SHR [30]. Susceptibility of angiotensin II was increased in the primary ventricle cardiomyocyte in SHR compared to those of normotensive Wistar-Kyoto rats [31].
Stimulated cardiac apoptosis occurs in essential hypertension [24]. In 1998, Fortuño et al. [23] observed that long-term administration of losartan (20 mg/kg) was associated with normalization of apoptosis and no change in the expression of Bcl-2 in SHR. These results were similar with our data. In 2002, González et al. [31] suggested that angiotensin II may participate in stimulating cardiac apoptosis in hypertension patients. González et al. [31] published data on losartan treated patients, which showed that both cardiomyocyte and noncardiomyocyte apoptosis decreased in DNA end-labeling assays.
In our study, losartan decreased apoptosis in the LV at week 5. So, it is possible that long term use of losartan, blocking angiotensin II receptor type 1A, contributes to blood pressure normalization by decreasing Bax protein expression level in the LV. On the basis of this finding, it can be proposed that angiotensin II and its receptor may participate in the stimulation of Bax protein in the LV, which makes SHR more susceptible to apoptosis.
Connexin 43 protein was increased in the LV of our SHR model. In order to confirm cardiac function in LV, we investigated connexin 43 protein expression in LV. Connexin 43 is one of the most abundant gap junction channel isoform in cardiomyocyte which is responsible for intercellular communications for vascular tone, cardiac rhythm and transfer of small molecules. Increased protein expression level of connexin-43 is involved in synchronizing myocardium. It affected on the electrical and molecular signal propagation in cardiomyocytes [13].
Connexin 43 double knockout mice showed symptoms of obstructed right ventricular outflow tract, cardiac malformation and neonatal death [30]. The changes in connexin 43 is related to pathology in SHR. Losartan protects against an abnormal elevation in connexin 43 level because connexin 43 regulates smooth muscle depolarization and cardiac functions [31]. Losartan decreased connexin 43 protein levels in our study.
A normal level of kallikrein is required required to maintain systemic blood pressure as it is involved in the regulation of blood pressure by controlling the activation of bradykinin. The reninangiotensin system and the nitric oxide/cyclic guanoxine monophosphate system also regulate blood pressure. Kallikrein system controls systemic, local hemodynamics and vascular remodeling, and kallikrein level is reduced in hypertension patients [27]. Kallikrein mRNA levels were increased in the LV of our SHR model. mRNA level of kallikrein was decreased at week 5 after losartan treatment in our study.
Losartan also has effects on the kallikrein-kinin system [27]. The renin-angiotensin and kallikrein-kinin systems are intricately connected, and some of the cardioprotective effects of losartan (10 mg/kg) are abolished by blocking the bradykinin B2 receptor signaling [14].
We may suppose the mechanism of losartan as follows. Losartan decreases blood pressure and angiotensin II signaling pathways that may affect reduced inflammation and apoptosis. As a result, it helps cardiac protection and decreased LV hypertrophy.
The limitations of this study is that these data are from rats, and the physiology and pathology of rats are not completely the same as humans. Also, the sample size was relatively small.
In conclusion, after losartan treatment Bax, CCR-2, MCP-1, TGF-β1, PERK, connexin 43 proteins and kallikrein mRNA expressions were reduced in our SHR model. Losartan also decreased inflammation, apoptosis and downregulated kallikrein-kinin.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
The authors gratefully acknowledge the support provided by Basic Science Research Program through the National Research Foundation of Korea funded by Ministry of Education (NRF-2013R1A1A3004619, and NRF-2017R1D1A1B03030831).