A rare case of dysembryoplastic neuroepithelial tumor combined with encephalocraniocutaneous lipomatosis and intractable seizures
Article information
Abstract
Encephalocraniocutaneous lipomatosis (ECCL) is a rare neurocutaneous syndrome that affects ectomesodermal tissues (skin, eyes, adipose tissue, and brain). The neurologic manifestations associated with ECCL are various including seizures. However, ECCL patients very rarely develop brain tumors that originate from the neuroepithelium. This is the first described case of ECCL in combination with dysembryoplastic neuroepithelial tumor (DNET) that presented with intractable seizures. A 7-year-old girl was admitted to our center because of ECCL and associated uncontrolled seizures. She was born with right anophthalmia and lipomatosis in the right temporal area and endured right temporal lipoma excision at 3 years of age. Seizures began when she was 3 years old, but did not respond to multiple antiepileptic drugs. Brain magnetic resonance (MR) imaging performed at 8 and 10 years of age revealed an interval increase of multifocal hyperintense lesions in the basal ganglia, thalamus, cerebellum, periventricular white matter, and, especially, the right temporal area. A nodular mass near the right hippocampus demonstrated the absence of N-acetylaspartate decrease on brain MR spectroscopy and mildly increased methionine uptake on brain positron emission tomography, suggesting low-grade tumor. Twenty-four-hour video electroencephalographic monitoring also indicated seizures originating from the right temporal area. Right temporal lobectomy was performed without complications, and the nodular lesion was pathologically identified as DNET. The patient has been seizure-free for 14 months since surgery. Although ECCL-associated brain tumors are very rare, careful follow-up imaging and surgical resection is recommended for patients with intractable seizures.
Introduction
Encephalocraniocutaneous lipomatosis (ECCL) is a rare congenital neurocutaneous syndrome of unknown etiology that was first described by Haberland and Perou in 19701). ECCL is characterized by unilateral cerebral malformations and ipsilateral lipomatous lesions involving the scalp, face, and eyes2). ECCL demonstrates a broad spectrum of clinical features. Diverse central nervous system anomalies in ECCL reportedly include intracranial and spinal lipomas, arachnoid cysts, asymmetric cerebral atrophy, various cysts, hydrocephalus, cortical dysplasia and vessel anomalies3). Brain tumor is also very rarely associated with ECCL. There were only 2 previous case reports on ECCL in combination with brain tumor. One report described a patient with a papillary glioneuronal tumor4), and the other described 2 unrelated patients with low-grade astrocytoma5). Dysembryoplastic neuroepithelial tumor (DNET) is a glioneuronal tumor according to the World Health Organization (WHO) classification6), which is characterized according to its benign nature, typical localization in the temporal lobe, and association with drug-resistant partial epilepsy in late childhood7). Here, we report a child affected by ECCL who presented with intractable seizures and DNET.
Case report
A 7-year-old girl was transferred to the Department of Pediatrics at the Asan Medical Center. Her major complaints included uncontrolled seizures and mild mental retardation. She was born with right anophthalmia with an orbital calcified mass, alopecia areata, and lipomatosis in the right temporal area. At another medical center, she underwent right temporal lipoma excision and eyeball reconstruction at 2 years of age. The first episode of complex partial seizure was documented when she was 3 years old, and she showed left-side tonic postures during this episode. Outside brain magnetic resonance imaging (MRI) at 2 and 6 years of age showed only lipomatosis on the right temporal bone, scalp, and cerebellopontine angle without definite intracerebral lesions (Fig. 1A–E). Her seizures were refractory to multiple antiepileptic drugs, including oxcarbazepine, levetiracetam, and clobazam. Brain MRI at 8 years of age revealed nonspecific hyperintense lesions in the basal ganglia, thalamus, and right temporal and left cerebral white matter in T2-weighted images (Fig. 1F–H), which was thought to be associated findings with ECCL. Multiple antiepileptic drugs, including topiramate and zonisamide, were added to control seizures, but she still had 3–4 seizures per month. At the age of 10 years, we performed further evaluations and considered epilepsy surgery. Repeated brain MRI revealed the interval increase of multifocal hyperintense lesions in the basal ganglia, thalamus, cerebellum, periventricular white matter, and especially the right temporal area (Fig. 1I–K). The nodular mass near the right hippocampus demonstrated the absence of N-acetylaspartate decrease on brain magnetic resonance spectroscopy but mildly increased methionine uptake on brain positron emission tomography, which is suggestive of a low-grade tumor (Fig. 2). Over 3 days of continuous electroencephalography (EEG)/video monitoring, 7 complex partial seizures were captured. Seizures were motionless staring or staring with bilateral tonic arms followed by turning the head to the right, and all ictal EEG recordings began with the runs of theta or delta activities from the right frontotemporal area; these findings support the right temporal origin of the seizures (Fig. 3). We decided surgery for uncontrolled seizures targeting putative right temporal low grade glioma, although she had multifocal intracranial lesions. Right anterior mesial temporal resection with tumor removal was performed without complications (Fig. 4), and the nodular lesion was pathologically identified as a DNET (Fig. 5) accompanied with right hippocampal sclerosis. The patient has been seizure-free for 16 months after surgery, although she is receiving oxcarbazepine, zonisamide, and clobazam.

Serial follow-up brain magnetic resonance imaging examination. (A, B) At 2 years and (C–E) 6 years of age, lipomatosis involving right temporal scalp and bone without intracranial lesion. (F–H) At 8 years of age, multiple nonspecific hyperintense lesions were documented in the basal ganglia in T2-weighted images. (I–K) White matter lesions (arrow) and a small mass (5 mm) are shown next to the right hippocampus (F and H circles), and the right temporal mass (I–K circles) had increased in size (11 mm) by 10 years of age.
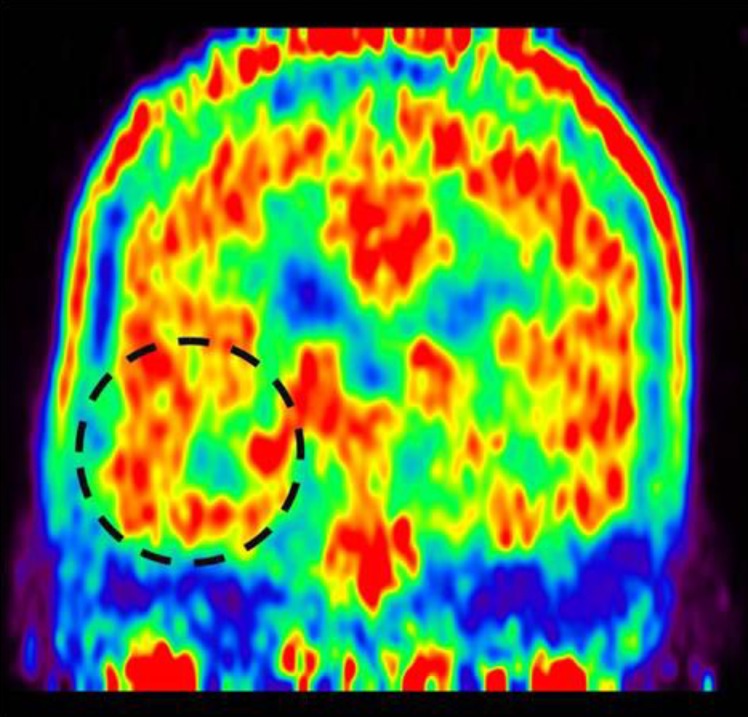
Methionine positron emission tomography showing increased methionine uptake in the right medial hippocampal area.
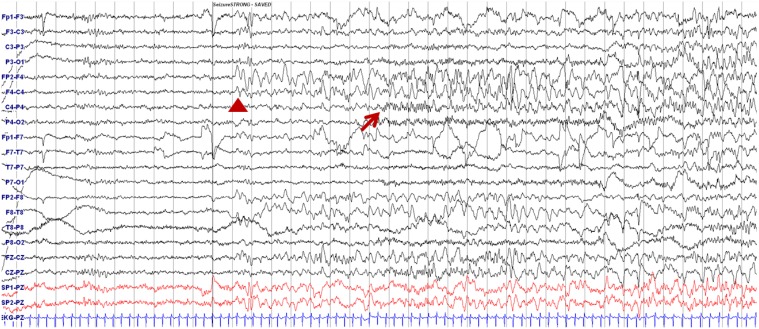
Electroencephalography showing ictal onset during seizure. Delta slowing over the right frontotemporal area (arrowhead) followed by runs of small-amplitude fast activities from the right centroparietal areas (arrow) are shown.
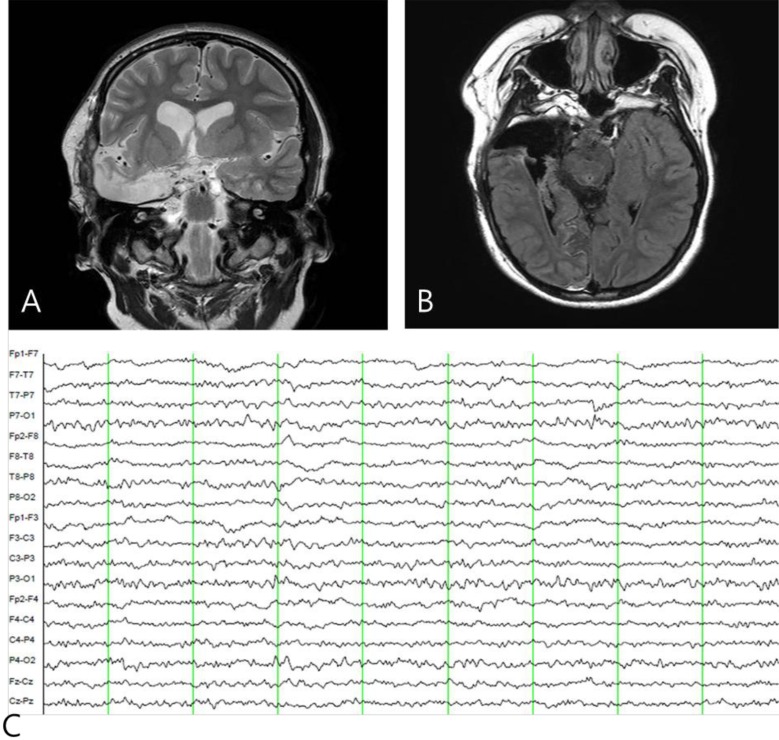
Postoperative brain magnetic resonance imaging (A, B) and electroencephalography (EEG) findings (C). (A, B) A right anterior temporal lobectomy was performed and (C) postictal EEG shows the disappearance of interictal epileptiform activity in the right temporal area.
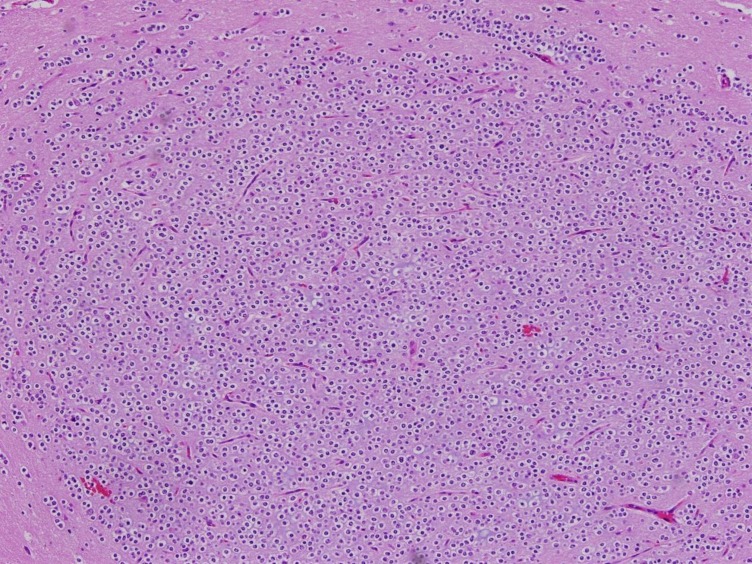
Histopathology of the excised dysembryoplastic neuroepithelial tumor (H&E, ×100).
Discussion
ECCL is a very rare congenital cutaneous syndrome that involves the mesoectodermal tissues, which are the neural crest derivatives such as the meninges, cranial vessels, dermis, hypodermis of the face and neck, connective tissue of head, and the dermal bones of the skull28). The clinical manifestations of ECCL include various skin anomalies, such as nevus psiloliparis, nonscarring alopecia, and subcutaneous lipoma. Ocular anomalies are diverse and include choristoma, corneal and other anterior chamber anomalies, ocular coloboma, and globe calcification. Intracranial and intraspinal lipomas are common central nervous system anomalies, and jaw tumor, multiple bone cysts, and aortic coarctation have also been observed in ECCL patients.
The differential diagnosis of ECCL includes Proteus syndrome, oculoectodermal syndrome, oculocerebrocutaneous syndrome, and epidermal nevus syndrome8). Proteus syndrome manifests as asymmetric body overgrowth, capillary malformation, connective tissue nevi, and disrupted adipose tissues, but generally demonstrates a progressive clinical course9). Oculoectodermal syndrome is a mild form of ECCL that presents with ocular and skin features10). Oculocerebrocutaneous syndrome is diagnosed by the typical characteristics of congenital malformations, including anophthalmia or microphthalmia, hypoplastic skin defects, skin appendages, and pathognomonic midhindbrain malformation11). Epidermal nevus syndromes are also included in a group of mosaic neurocutaneous conditions that are frequently associated with intracranial/intraspinal lipomas12). In accordance with the revised diagnostic criteria for ECCL8), the major diagnostic criteria includes ocular choristoma, nevus psiloliparis, intracranial or intraspinal lipoma, and jaw tumor, multiple bone cysts, or aortic coarctation. In addition, the involvement of more than 2 different systems is needed for the definitive diagnosis of ECCL8). In our patient, 2 different systems were involved with possible nevus psiloliparis plus nonscarring alopecia and a definitive diagnosis of ECCL was made.
The brain anomalies associated with ECCL can be divided into three groups: (1) the development of abnormal cell types, including intracranial and intraspinal lipomas; (2) congenital anomalies of the meninges; and (3) vascular defects3). Cerebral hemispheric asymmetry, corpus callosum dysgenesis, and cortical dysplasia are reportedly common findings in the brains of ECCL patients3). However, concomitant brain tumors are alleged to develop rarely in ECCL patients. As far as we know, only 2 reports of pediatric patients have been published. One patient with papillary glioneuronal tumor was previously reported in an ECCL patient who presented with paraplegia4). The other 2 patients were diagnosed with ECCL, low-grade astrocytoma, and seizure5). Our patient is the first case of ECCL in combination with DNET to be reported.
Considering the very few reported cases of ECCL8), the scarce reports of low grade tumor in ECCL may not be unusual but rather the low grade tumors can be associated with ECCL. The etiology and pathogenesis of ECCL has not been clearly identified. ECCL sporadically occurs involving the skin and bone, and some suggest that it is a mosaic condition8) or somatic mutation like Sturge-Weber syndrome13). Previous reports suggested that several genes like NF1 gene, genes of RAS-MAPK pathway or genes involving vasculogenesis might be associated28). ECCL is also frequently associated with developmental brain anomalies such as focal cortical malformation3), which is frequently associated with DNET14). According to WHO, DNET is a grade I glial tumor that predominantly involves the temporal lobe and accounts for approximately 60% of cerebral lesions15). Its benign clinical course and stability mostly delays the detection of the mass after the initial seizure symptoms develop7), and DNET is usually revealed by uncontrolled partial seizures in young patients. The epileptogenic mechanisms of DNET remain unknown, and associated focal cortical dysplasia could play a role in the epileptogenicity of DNET7). Though the pathogenesis of ECCL is not fully understood, the development of abnormal cell type and frequent association with cortical dysplasia in ECCL may prone to develop the DNET.
In our patient, it took three years to identify intracranial lesions after initial seizure treatment, and the later identified DNET was associated with pathologically proven hippocampal sclerosis. Seizures were completely controllable after tumor removal and hippocampal resection. This is a very rare case of DNET associated with ECCL, a congenital rare neurocutaneous syndrome in a child having intractable seizures, which suggests a common developmental pathophysiology of these diseases. Careful monitoring and follow-up brain imaging is also needed in patients with ECCL as in other neurocutaneous syndrome and epilepsy surgery can be considered as another treatment option in patient with ECCL and intractable seizure.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.