A novel mutation of ABCC8 gene in a patient with diazoxide-unresponsive congenital hyperinsulinism
Article information
Abstract
Congenital hyperinsulinism (CHI) is a rare condition that can cause irreversible brain damage during the neonatal period owing to the associated hypoglycemia. Hypoglycemia in CHI occurs secondary to the dysregulation of insulin secretion. CHI has been established as a genetic disorder of islet-cell hyperplasia, associated with a mutation of the ABCC8 or KCNJ11 genes, which encode the sulfonylurea receptor 1 and the inward rectifying potassium channel (Kir6.2) subunit of the ATP-sensitive potassium channel, respectively. We report the case of a female newborn infant who presented with repetitive seizures and episodes of apnea after birth, because of hypoglycemia. Investigations revealed hypoglycemia with hyperinsulinemia, but no ketone bodies, and a low level of free fatty acids. High dose glucose infusion, enteral feeding, and medications could not maintain the patient's serum glucose level. Genetic testing revealed a new variation of ABCC8 mutation. Therefore, we report this case of CHI caused by a novel mutation of ABCC8 in a half-Korean newborn infant with diazoxide-unresponsive hyperinsulinemic hypoglycemia.
Introduction
Neonatal hypoglycemia is caused by numerous clinical conditions, such as birth asphyxia, small for gestational age (SGA), premature birth, and infants born to diabetic mothers, systemic disorders, hormonal disorders or fatty acid oxidation disorders. Of them, infants that experience birth asphyxia, SGA, premature birth, and born to diabetic mothers usually have transient hyperinsulinism that resolves quickly, even though it may be quite severe1).
However, persistent hypoglycemia can be caused by hyperinsulinism, even if there is no history or biochemical evidence of maternal diabetes, birth asphyxia, premature birth or SGA. Congenital hyperinsulinism (CHI or persistent hyperinsulinemic hypoglycemia of infancy; OMIM#256450) is the most common cause of nonketotic persistent severe hypoglycemia in neonates and caused by inappropriate insulin secretion from pancreatic β cells23). Inherited or sporadic mutations in the regulation of the potassium channel involved in insulin secretion by the pancreatic β cells can lead to CHI1). The most common causes of CHI are mutations that inactivate the ABCC8 and KCNJ11 genes encoding the sulfonylurea receptor 1 (SUR1) and the inward rectifying potassium channel (Kir6.2) subunit of the ATP-sensitive potassium channel4). It is rare but critical condition that occurs during the neonatal period and is accompanied by hypoglycemia. Persistent hypoglycemia is a major risk factor for mental retardation, therefore, early diagnosis and treatment are extremely important25).
Recently, we examined a baby with diazoxide unresponsive CHI with a novel mutation in the ABCC8 gene and would like to report our case in this paper.
Case report
A 2-day-old girl was transferred to our hospital with repetitive myoclonic movements and apnea. She was born at 41 weeks of gestation and her birth weight was 4.1 kg (90th–97th percentile). Mother is a Chinese and father is a Korean. Father denied any perinatal problems such as gestational diabetes mellitus, prolonged rupture of membrane, polyhydramnios or oligohydramnios. The patient was delivered vaginally and her Apgar scores were 9 at 1 minute and 10 at 5 minutes after birth, respectively. She was transferred to our hospital at 2 days of age because she presented frequent myoclonic movements of the upper and lower limbs and oxygen desturation around 70% after birth.
On admission, blood pressure was 69/41 mmHg, heart rate was 158 beats per minute, respiration rate was 50 breaths per minute and body temperature was 36.7℃. Weight was 3.8 kg (75th–90th percentile), length was 53.5 cm (90-95 percentile), head circumference was 34 cm (25th–50th percentile), chest circumference was 34.5 cm, and abdominal circumference was 35 cm. She had a symmetric appearance and did not have any congenital malformations.
She was lethargic and presented with hypotonic posture. Upon physical examination, she showed clear breathing sounds, regular heart beat and no cardiac murmur. She had a soft abdomen, active bowel sounds and no hepatosplenomegaly. After she was admitted, she displayed repetitive myoclonic movements of upper and lower limbs, apnea and oxygen desaturation. A blood sugar test (BST) was performed immediately and the result was 18 mg/dL. Eight milliliters of 10% dextrose solution was injected as a bolus and 10% dextrose solution was continuously infused with a glucose infusion rate (GIR) of 4.07 mg/kg/min. Phenobarbital was injected concomitantly. BST was performed every 30 minutes after the bolus infusion of dextrose. The GIR was increased up to 20.8 mg/kg/min steadily and bood sugar was elevated to 95 mg/dL. After the blood sugar level reached a normal range, she stopped experiencing myoclonic seizure and apnea.
Initial blood work showed a hemoglobin of 18.4 g/dL, hematocrit of 55%, a white blood cell count of 24,830/mm3, and a platelet count of 283,000/mm3. Serum blood glucose was 2 mg/dL on initial chemistry and the BST revealed a blood glucose of 18 mg/dL. Serum sodium level was 141.3 mmol/L, potassium was 5.1 mmol/L and chloride was 104.4 mmonl/L. Serum calcium was 8.8 mg/dL, bicarbonate was 19 mmol/L, and ammonia was 69 µg/dL (reference range, 19–87 µg/dL). C-reactive protein was 4.6 mg/dL (reference range, 0–5 mg/dL) and no bacterial growth was noted on blood culture. Laboratory findings showed no ketone bodies in serum and urine, insulin was 9.1 mIU/L (reference range, 2.0–25.0 mIU/L), and C-peptide was 5.96 ng/mL (reference range, 1.1–4.4 ng/mL) when serum glucose level was 13 mg/dL. The free fatty acids level was 124 µEq/L (reference range, 172–586 µEq/L).
Chest X-ray showed no ground glass opacity. On the second day in the hospital, brain magnetic resonance image with diffusion was performed because of repetitive seizures and apnea. High signal intense lesions in left parietal lobe, both occipital cortices, and subcortical white matter were observed (Fig. 1). On day 4 of hospital stay, abdominal ultrasonography was performed to determine the presence of any pancreatic lesions such as adenoma, but no pancreatic problems could be detected. Electroencephalogram showed some attenuation of the background and an echocardiogram showed a hypertrophied interventricular septum (5.9 mm, >97th percentile) and a left ventricular posterior wall (4–5 mm, upper normal limit) without obstruction of the left ventricular outflow tract. Computed tomography and positron emission tomography of the pancreas were not performed because of the risk of radiation exposure.

Diffusion-weighted magnetic resonance imaging of the brain performed on hospital day 2 because of frequent seizures, showing high-intensity signal lesions in the left parietal cortex, both occipital lobe cortices, and subcortical white matter.
We fed her using Levin tube because she could not suck and swallow milk well on the 1st day of admission. When feeding volume reached to 150 mL/kg, blood glucose levels fluctuated between 31 to 162 mg/dL under the high intravenous GIR of 11.06–16.57 mg/kg/min. We started corticosteroid treatment but blood glucose was not stable. We prescribed diazoxide on day 10. However, blood glucose was still fluctuated with 20 mg/kg/day of diazoxide, so we added octreotide subcutaneously 21 days after being admitted (Fig. 2).
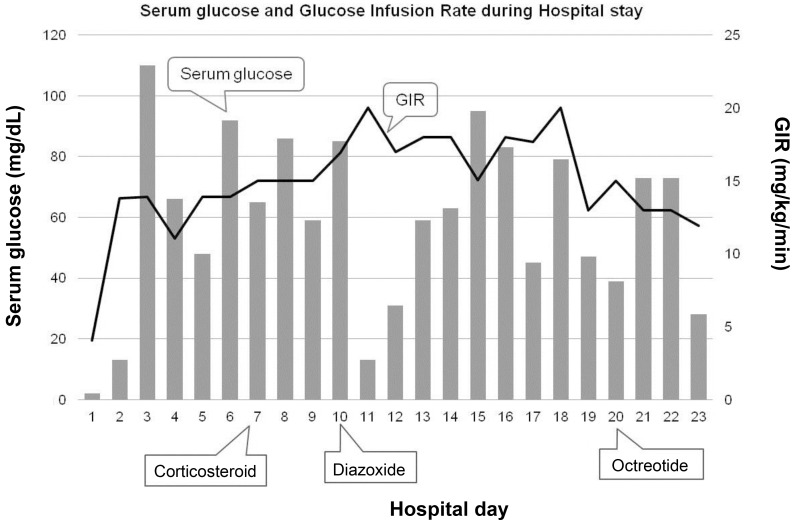
Serum glucose levels and glucose infusion rate (GIR) during hospital stay. The level of serum glucose fluctuated despite the high GIR and medication. Gray bars indicate serum glucose levels; solid line indicates GIR.
Full sequencing of the ABCC8 gene was performed because ABCC8 mutation is more prevalent than KCNJ11 in Korea2). Genetic analysis revealed a new heterozygotic variation of c.4608G>A on exon 38 of ABCC8 (Fig. 3). On the basis of her clinical and laboratory findings, the variation seems to be a reason of her CHI.
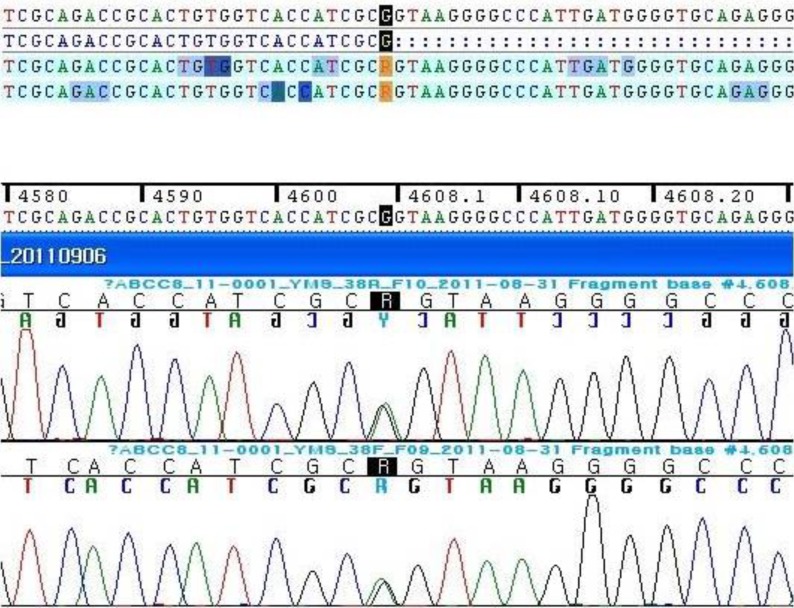
Genetic analysis revealed a heterozygote variation of c.4608G>A on exon 38 of ABCC8.
After the genetic diagnosis, father wanted her to be transferred to other hospital when she was 24 days old. At that time, blood glucose was still fluctuating even though she was receiving diazoxide and octretide injections.
She has not visited our hospital after discharge, but we received a report on her clinical condition from a relative in Korea 3 years later. She is living in China and maintains a euglycemic state without medicine or surgery. She developed well but she has been suffering from intermittent seizures.
Discussion
Up to now, there have been many nation-wide studies about ABCC8 and KCNJ11 mutations in patients with CHI3678) and more than 150 mutations have been reported in the ABCC8 gene9). Twelve different mutations of the ABCC8 gene have been reported in Korean patients2).
Here we report on a new mutation in the ABCC8 gene, a single base alteration at a position 4608 (4608G>A) on exon 38. The alteration was neither identified in the polymorphism and mutation database until now (http://www.ncbi.nlm.nih.gov/snp). The site of the new alteration is the last base sequence of exon 38 in the ABCC8 gene and might affect exon-intron splicing.
The human ABCC8 gene located on chromosome 11p15.1 has 39 exons and encodes the 1581 amino acids of the SUR1 protein. The SUR1 protein is one subunit of the ATP sensitive KATP channels in pancreatic β cells. The pancreatic KATP channel plays a major role in regulating insulin secretion from the β cell. A defective or inactivated pancreatic KATP channel can cause hyperinsulinemia despite hypoglycemia10).
To diagnose hyperinsulinemic hypoglycemia, clinicians rely on clinical and laboratory findings at the moment of hypoglycemia. Hyperinsulinemia can be supported by inappropriately high insulin concentrations accompanied by hypoglycemia, low or no fatty acids and ketone bodies, a glycemic response to glucagon injection and a high GIR to maintain euglycemia11). In our case, CHI was diagnosed based on GIR of 20.8 mg/kg/min required to maintain euglycemia, 9.1 mIU/L of insulin and no ketone bodies with a serum glucose level of 13 mg/dL, and low level of free fatty acids.
Treatment for CHI requires multidisciplinary apporaches including nutrition, medicine and surgery511). The goal of CHI treatment is to preventing brain damage, however, it is one of the most difficult diseases to manage. The nutritional approach includes hypertonic glucose infusion and enteral feeding. Medical treatment includes administration of diazoxide, octreotide, nifedipine and glucagon. Diazoxide is a benzothiazine derivative that acts on the SUR1 subunit of the KATP channel. It keeps the channel open and stops the secretion of insulin. Octreotide is a somatostatin analogue that inhibits insulin secretion by activating somatostatin receptor 5. Nifedipine is a calcium channel blocker and has been reported as effective drug in regulating insulin secretion1213). Glucagon stimulates glycogenolysis and gluconeogenesis. But it can cause paradoxical insulin secretion. When CHI patients fail to respond to medical and nutritional treatment, a pancreatectomy should be considered. The extent of surgery depends on CHI subtype, diffuse and focal. However, as pancreatectomy has several risks including the inherent risks of surgery, persistent hypoglycemia and risk of developing diabetes mellitus postoperatively5), so surgery is carefully considered.
In this case, serum glucose levels fluctuated in spite of a GIR of 20.8 mg/kg/min and enteral feeding. We tried corticosteroids, diazoxide and octreotide, but those were ineffective in maintaining normal blood glucose levels. Although treatment of CHI is primarily based on the use of diazoxide, CHI caused by recessive inactivating mutations in the ABCC8 and KCNJ11 genes is usually unresponsive to medical treatment with diazoxide10). After being transferred to another hospital, we heard that she slowly responded to medical managements over a period of 2 months and she finally could be taken off diazoxide and octreotid at around 3 months of age. We cautiously hypothesize that it is possible that she experienced spontaneous remission of CHI. According to previous reports, diffuse and focal CHI can resolve spontaneously over time214). Apoptotic death of insulin-oversecreting β cells and functional shutdown of insulin secretion were suggested as possible mechanisms for the spontaneous remission of CHI11).
Though only a single base was altered, c.4608G>A on exon 38 of ABCC8 in this case might be a possible cause of CHI, along with her symptoms and laboratory findings. The genetic of CHI is known as recessive trait, however, the single alteration of our case might be overdominant15). Functional studies and genetic analyses of parents are necessary to confirm. In the aspect of neurologic complication of seizure disorder in this patient, early diagnosis and aggressive nutritional and medical management were important in preventing permanent brain damage.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.