Atypical hemolytic uremic syndrome and eculizumab therapy in children
Article information
Abstract
Hemolytic uremic syndrome (HUS) is often encountered in children with acute kidney injury. Besides the well-known shiga toxin-producing Escherichia coli-associated HUS, atypical HUS (aHUS) caused by genetic complement dysregulation has been studied recently. aHUS is a rare, chronic, and devastating disorder that progressively damages systemic organs, resulting in stroke, end-stage renal disease, and death. The traditional treatment for aHUS is mainly plasmapheresis or plasma infusion; however, many children with aHUS will progress to chronic kidney disease despite plasma therapy. Eculizumab is a newly developed biologic that blocks the terminal complement pathway and has been successfully used in the treatment of aHUS. Currently, several guidelines for aHUS, including the Korean guideline, recommend eculizumab as the first-line therapy in children with aHUS. Moreover, life-long eculizumab therapy is generally recommended. Further studies on discontinuation of eculizumab are needed.
Introduction
Hemolytic uremic syndrome (HUS) is the common cause of acute kidney injury in children. It is induced by various clinical situations such as several infections, medications, metabolic disease, bone marrow transplantation, and genetic abnormalities in the complement and coagulation pathway. HUS is characterized by the clinical triad of microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and acute kidney injury, and belongs to the subgroup of thrombotic microangiopathy (TMA).123)
Shiga toxin-producing Escherichia coli (STEC) is a famous bacterium causing HUS, called STEC HUS, which typically presents after hemorrhagic gastroenteritis. STEC HUS is the most common form of HUS in children. Among the other causes of HUS, complement-mediated genetic HUS, also called as atypical HUS (aHUS), is one of the most important causes and has been extensively studied recently. Thrombotic thrombocytopenic purpura (TTP) is an acquired or congenital disease caused by a severe deficiency (generally below 10%) in plasma ADAMTS13 activity.4) TTP is no longer classified as a variant of aHUS; however, clinicians have to distinguish between TTP and aHUS. This review does not include more details on TTP.
Eculizumab is a newly developed drug for blocking the terminal complement pathway, which is generally used in paroxysmal nocturnal hemoglobinuria and aHUS. In this review, aHUS, eculizumab, and the Korean guideline for aHUS will be discussed.
What is aHUS?
aHUS is a rare, chronic, and devastating disorder caused by uncontrolled complement activation leading to endothelial injury, platelet aggregation, MAHA, and progressive damages to vital organs, leading to stroke, heart attack, renal failure, and death. Initially, aHUS meant any HUS not caused by STEC. However, as several clinical conditions other than STEC, such as pneumococcal infection, cobalamin deficiency, autoimmune disease, and bone marrow transplantation, have been known to lead to HUS, the term aHUS is generally used these days to define HUS without coexisting disease or specific infection, which is mostly a disease of complement alternative pathway overactivation.56) Furthermore, mutations in coagulation pathway genes other than the complement pathway genes have been found to be associated with aHUS. Thus, aHUS can be divided into several groups according to its etiology, including mutations in the complement alternative pathway gene, anticomplement factor H autoantibody, mutations in coagulation pathway genes, and combinations thereof.7)
Historically, several genes causing aHUS had been identified, including complement factor H (CFH), complement factor B (CFB), complement factor I (CFI), complement 3 (C3), and membrane cofactor protein (MCP). As mentioned before, mutations in coagulation pathway genes such as thrombomodulin (THBD), diacylglycerol kinase-ε (DGKE), and plasminogen (PLG) can induce aHUS.7) Generally, genetic abnormalities in aHUS have been detected in about 60% of patients.1) Anti-CFH antibody production results in an acquired form of aHUS, the pathogenesis of which is a kind of autoimmune disease. Interestingly, production of this autoantibody is associated with genetic susceptibility. Anti-CFH antibody is frequently produced in association with homozygous deletion of CFH-related protein 1 gene (CFHR1), which encodes CFH-related protein 1.8) Additionally, 2 cases of anti-CFI antibody in aHUS have been reported; however, its clinical significance needs further investigation.9)
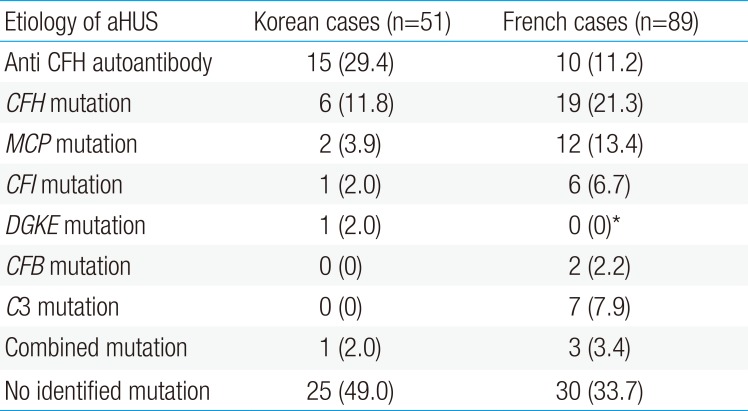
Recently, a Korean pediatric series of aHUS was published, which investigated 51 patients with aHUS and reported the results of genetic testing. From this multicenter cohort, 15 patients (29.4%) had anti-CFH antibody, which is the most common cause of aHUS. However, in 49% of patients, the investigators failed to find specific genetic mutations. Among the detected mutations, mutations in CFH were the most common (6 of 11 patients, 54.5%). A comparison of etiology in pediatric aHUS between a Korean and a French cohort study, which is one of the largest studies worldwide, is shown in Table 1; however, the French cohort did not include DGKE mutation.57)

Comparison of etiology of pediatric aHUS between Korean and French cohorts
Although mortality and renal outcomes in aHUS vary according to the involvement of each gene, about 20% of children progressed to end-stage renal disease (ESRD) or died at the first episode or within 1 month after onset, and 30% within 1 year. Relapse occurred in approximately half of the patients who survived the first episode and did not reach ESRD.510) The mortality rate was significantly higher in children than in adult patients.5) Children having CFH mutations demonstrated the most severe manifestations and outcomes, with a risk of death or ESRD of about 30% at the first episode. On the other hand, children with MCP mutations had the best prognosis, with a long-term risk of ESRD of 25% at a median follow-up of 17.8 years despite frequent relapses.511)
Although TMA processes are induced mainly in the kidney, other organs can be involved. That is, aHUS can affect multiple vital organs and tissues besides the kidneys, such as the cardiovascular, gastrointestinal, pulmonary, visual, and central nervous systems. The most common extrarenal involvements during the acute stage is central nervous system involvements, with symptoms such as seizure, decreased mentality, visual problems, and comatose. In a large aHUS registry study in Turkey published in 2017, 42% (61 of 146) of patients had extrarenal manifestations.12) aHUS is known to be a microvascular disease. However, macrovascular symptoms including cerebral artery stenosis and peripheral gangrene have been reported.13)
When should aHUS be suspected? How can aHUS be diagnosed?
When children with the clinical triad of MAHA, thrombocytopenia, and acute kidney injury present to the clinic, HUS can be easily diagnosed. Next, differential diagnosis for HUS has very important clinical implications for evaluation, prognosis, and treatment. Through medical history taking and physical examination, HUS secondary to a coexisting condition such as bone marrow transplantation, pneumococcal infection, and other infections can be usually eliminated. Pneumococcal HUS mostly presents with symptoms of invasive infection, including pneumonia with empyema, meningitis, and bacteremia in young children aged <2 years.14) TTP, which is another group in TMA, can be excluded according to ADMATS13 activity, and cobalamin deficiency can be also ruled out on the basis of serum homocysteine and methionine levels. Blood samples for ADMATS13 activity should be taken before the initiation of plasma therapy.
STEC infection should be excluded as soon as possible in a patient with HUS by means of culture procedures or real-time polymerase chain reaction. Negative results are observed in about 30% of patients who had been clinically classified as having STEC HUS, frequently owing to delayed stool collection.15) Therefore, it sometimes becomes difficult to diagnose or exclude STEC HUS. Typically, about 80% of children with STEC HUS have bloody diarrhea and STEC HUS accounts for most TMA cases in children. Therefore, STEC HUS should be preferentially presumed in children with common gastrointestinal symptoms.16)
aHUS can be suspected in HUS patients without a history of diarrhea or bloody stool. However, even if children with HUS have gastrointestinal symptoms, aHUS other than STEC HUS should be suspected in the presence of the following conditions: very early onset age (<6 months), no definite onset time, recurrent or familial HUS, and low C3 level.17)
Genetic testing for complement abnormalities is recommended for aHUS patients especially in those with relapse, familial history and de novo posttransplantation HUS, as well as in those scheduled for renal transplantation for aHUS.1718) Additionally, confirming aHUS requires genetic testing; however, failing to find causative genetic mutations does not exclude the diagnosis of aHUS because specific genetic mutations still could not be detected in 40%–50% of aHUS patients.
What are the traditional treatments for aHUS?
1. Plasma therapy
Plasma therapy (plasma exchange or plasma infusion) has been traditionally conducted as first-line treatment in children suspected of having aHUS after the study published by Bell et al.19) in 1991, although TTP and aHUS were not distinguished in this report. Theoretically, plasma exchange can provide large amounts of nonmutated complement factors and remove the autoantibody or mutated factors that exist in plasma. Therefore, urgent plasma exchange is generally recommended in cases suspected of aHUS. However, plasma exchange is a remarkably specialized and complicated technique. A recent audit for the European guidelines in 2009 demonstrated that 31% of 51 children who received plasma therapy experienced catheter-related complications such as hemorrhage, chylothorax, and thrombosis.20) Additionally, this study showing a low rate of plasma exchange in infants younger than 6 months reminds of the technical difficulties with a lack of expertise and equipment, especially in small infants. Sensitization to the plasma component due to massive transfusion was also observed in 8 patients.
Plasma therapy may achieve hematologic remission; however, generally, it does not result in a significant improvement of renal function, which deteriorates the quality of life.20) Noris et al.11) reported that although plasma therapy induced complete or partial remission in about 80% of acute episodes of aHUS in children, half of the patients had died or progressed to ESRD at 3-year follow-up. The rate of progression to ESRD during the first episode of aHUS was similar between children with CFH mutations who were treated with intense plasma exchange and those who were not.5) Plasma exchange is usually performed with an exchange of 1.5 plasma volumes per session, and fresh frozen plasma is used for replacement. Plasma infusion (10–20 mL/kg) should be considered if plasma exchange is not available. Plasma therapy is continued daily until the disease activity is controlled from 5 days to 2 weeks. Thereafter, the subsequent frequency is 5 times per week for 2 weeks and then 3 times per week for 2 weeks. Patients receive plasma therapy every 2–4 weeks as long-term maintenance treatment.17)
2. Liver transplantation
In children with aHUS caused by a specific mutation in the complement alternative pathway, including CFH, CFI, CFB, and C3, isolated liver transplantation or combined liver and kidney transplantation can be a therapeutic option, as the liver is the major organ producing these factors.17) In these cases, isolated kidney transplantation can neither cure aHUS nor prevent its recurrence. There have been several case reports on liver transplantation with or without kidney transplantation, and also a few in Korea. CFH mutation caused aHUS in 2 young children, and liver transplantation improved the plasma CFH level and prevented recurrence, although one patient died of an infectious complication.2122) However, the optimal timing of liver transplantation in children with aHUS is still uncertain.
3. Kidney transplantation
Kidney transplantation alone cannot cure aHUS but plays a role of renal replacement therapy. In addition, without clarifying genetic causes and providing appropriate treatment, aHUS is likely to recur in the transplanted kidney, although the risk of recurrence varies according to the genetic abnormalities. The overall rate of aHUS recurrence after kidney transplantation is 60% in the preeculizumab era and renal survival was 30% at 5-year follow-up.23) Living donor kidney transplantation is contraindicated in aHUS because of the high risk of recurrence. Prophylactic plasma therapy has been recommended for posttransplantation recurrence and, recently, prophylactic eculizumab has been suggested.17)
What is eculizumab? How effective is it in aHUS?
Eculizumab is a humanized monoclonal anti-C5 antibody that inhibits C5 cleavage to C5a and C5b, and ultimately prevents the formation of the membrane attack complex. Therefore, the terminal complement pathway could be blocked (Fig. 1).24) It has been successfully used in paroxysmal nocturnal hemoglobinuria for more than 10 years.25) Theoretically, blocking the terminal complement pathway by using eculizumab can improve aHUS, the pathogenesis of which is based on uncontrolled complement activation; moreover, eculizumab can reduce inflammation, endothelial damage, thrombosis, and kidney injury.26) Several clinical trials from Alexion Pharmaceuticals have shown the efficacy of eculizumab in inducing hematologic normalization, and in improving renal function in adults and children with aHUS.272829) A recent prospective trial conducted in patients younger than 18 years demonstrated that eculizumab achieved complete TMA response, defined as hematologic normalization (platelet count ≥150×109/L, lactate dehydrogenase levels lower than the upper limit of normal) and renal improvement (≥25% decrease in serum creatinine level from baseline) in 64% of patients after 26 weeks, and was well tolerated. Moreover, most patients showed improvement in all hematologic and renal parameters.30) A retrospective study by Fakhouri et al.31) revealed that eculizumab was more effective than plasma therapy used in a historical control group. It is also important that early initiation of eculizumab (within 6 days) improves renal function better than a later initiation. When eculizumab is initiated as soon as possible after aHUS manifestation, greater improvement of renal function can be expected.28)
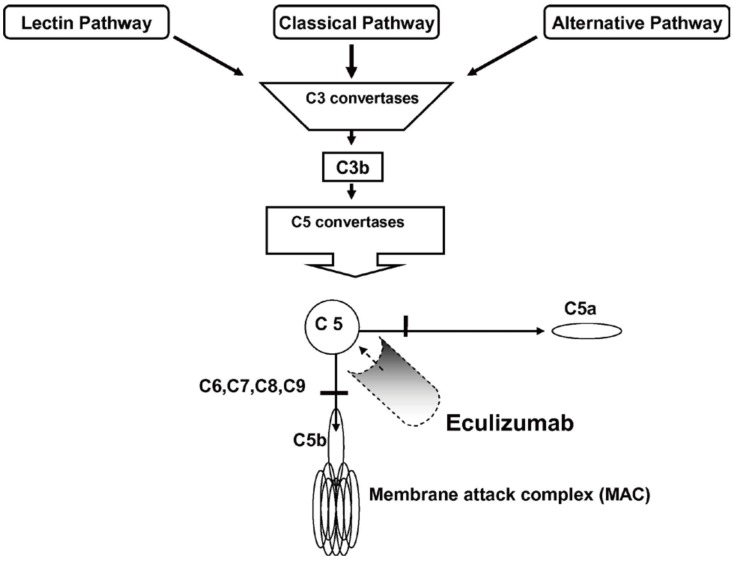
Blockade of terminal complement activation by eculizumab. There are 3 known pathways for initial complement activation: classical, alternative, and lectin pathway. These 3 pathways converge at the point of C3 activation. The final pathway starts with the formation of C5 convertase. Eculizumab inhibits terminal complement activation by binding to C5 and preventing the formation of membrane attack complex. Reprinted from Brodsky. Blood reviews 2008;22:65–74, with permission from Elsevier.24)
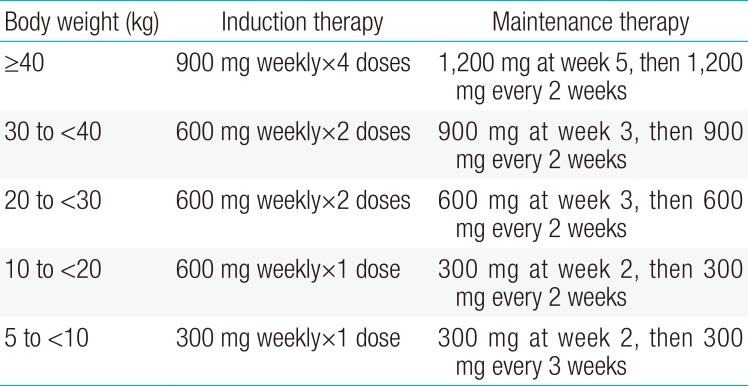
Eculizumab is now officially accepted for the treatment of aHUS in many countries, including European countries, the United States, and Japan. In Korea, very few children with aHUS are treated with eculizumab, although the process of setting health insurance details between pharmaceuticals and the government is yet unfinished. Eculizumab is administered as an intravenous infusion, and the recommended dose and treatment schedule in children with aHUS from the product label is shown in Table 2.

Recommended dose and treatment schedule of eculizumab in children with atypical hemolytic uremic syndrome
All guidelines recommend urgent eculizumab, if available, as initial therapy, and lifelong eculizumab therapy is generally recommended, especially for kidney transplant recipients with CFH mutation and for those with a <20 mL/min/1.73 m2 glomerular filtration rate.161718) Several trials for the discontinuation of eculizumab have been conducted because lifelong therapy has some problems, including potent serious meningococcal infection, drug adverse reaction, recurrent vascular access, and high cost. However, discontinuation of eculizumab may increase the risk of further clinical manifestation of aHUS and is currently not supported by evidence.3233) On the contrary, some authors suggested careful discontinuation with aggressive home monitoring for early detection of relapse, especially in cases with MCP mutations, homozygous CFHR3/R1 deletions, anti-CFH antibodies, no identifiable mutations, and CFI mutations.3435)
The major adverse effect of eculizumab is meningococcal infection because defense against this bacterium depends on the lytic terminal complement complex, which is blocked by eculizumab. Therefore, prevention through vaccination and antibiotic administration is essential.18) All routine vaccinations including live vaccines in children receiving eculizumab can be done as in primary complement deficiency or asplenia.36)
What does the recent Korean guideline say?
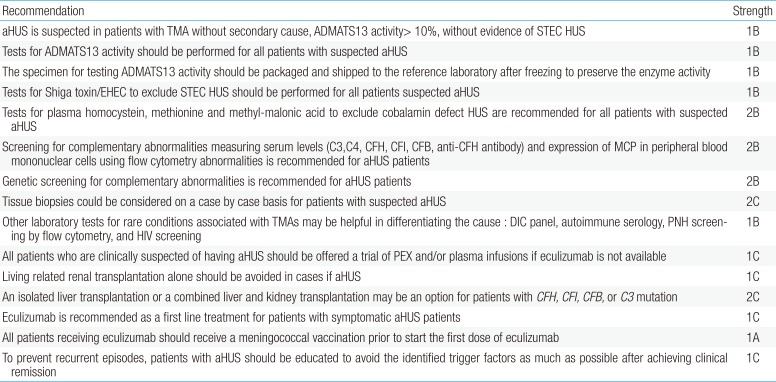
Recently, the Korean practical guidelines for aHUS was developed by the Korean aHUS Working Group, which is composed of physicians representing the Korean Society of Pediatric Nephrology, Korean Society of Nephrology, Korean Society of Hematology, and Korean Society on Thrombosis and Hemostasis. All recommendations in this guideline are listed in Table 3.17)

Recommendations in the Korean guideline for aHUS
Conclusions
aHUS is a critical disease that leads to irreversible organ damage, especially the kidney, if adequate treatment is not performed. Traditional treatments have not shown sufficient and lasting effectiveness. There are expectations for the newly developed eculizumab, and thus far, eculizumab is giving a lot of hope and further research is needed.
Notes
Conflict of interest: The authors declared no conflict of interest.