Prognostic factors and treatment of pediatric acute lymphoblastic leukemia
Article information
Abstract
The event-free survival (EFS) for pediatric acute lymphoblastic leukemia (ALL) has shown remarkable improvement in the past several decades. In Korea also, a recent study showed 10-year EFS of 78.5%. Much of the improved outcome for pediatric ALL stems from the accurate identification of prognostic factors, the designation of risk group based on these factors, and treatment of appropriate duration and intensity according to risk group, done within the setting of cooperative clinical trials. The schema of first-line therapy for ALL remains mostly unchanged, although many groups have now reported on the elimination of cranial irradiation in all patients with low rates of central nervous system relapse. Specific high risk subgroups, such as Philadelphia chromosome-positive (Ph+) ALL and infant ALL continue to have significantly lower survival than other ALL patients. The introduction of tyrosine kinase inhibitors into therapy has led to enhanced outcome for Ph+ ALL patients. Infant ALL patients, particularly those with MLL rearrangements, continue to have poor outcome, despite treatment intensification including allogeneic hematopoietic cell transplantation. Relapsed ALL is a leading cause of mortality in pediatric cancer. Recent advances in immunotherapy targeting the CD19 of the ALL blast have shown remarkable efficacy in some of these relapsed and refractory patients. With improved survival, much of the current focus is on decreasing the long-term toxicities of treatment.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common cancer in the pediatric age group and is responsible for the most cancer-related deaths in children and adolescents. In Korea, the age-standardized incidence rate of ALL is approximately 28 patients per million in the 0–14 year old age group1). Whereas 50 years ago the survival for pediatric ALL was 10%–20%2), currently long-term overall survival (OS) rates are 80%–90%345). In Korea also, we reported a 10-year event-free survival (EFS) of 78.5% and OS of 81.9% for a large number of patients treated on CMCP-ALLL2005 and -ALL2008 regimens6). Much of the improvement in survival stems from the classification of patients into risk groups based on prognostic factors, and the adjustment of treatment intensity according to risk group, within the setting of national and multinational clinical trials. Current focus is on further understanding the biology of the disease and developing novel therapeutics in order to salvage patients who relapse or remain refractory to first-line treatment, as well as on minimizing the long-term adverse effects of chemotherapy.
Prognostic factors
Well-established prognostic variables include patient factors such as age, initial presenting white blood cell (WBC) count, the genetic and immunophenotypic characteristics of the leukemic blast, and individual response to therapy.
1. Age and WBC count
In precursor B cell (Pre-B) ALL, patient age at diagnosis and presenting WBC count are independent prognostic factors. Patients diagnosed between the ages of 1 and 10 have a superior outcome compared with those <1 year old or ≥10 years old. Infant ALL is particularly known to have poor survival with a 4-year EFS of 47% according to a multinational study7). High WBC count at diagnosis is also an adverse factor, with patients presenting with WBC count≥50,000/mm3 having worse outcome. Age and WBC count at diagnosis are combined in the categorization of clinical risk group according to National Cancer Institute/Rome criteria, with patients between the ages of 1 and 9.99 years and having a WBC count<50,000/mm3 termed ‘standard risk,’ while the remainder are considered ‘high risk’8). In contrast to patients with Pre-B ALL, age and WBC count have less of a role in determining prognosis in patients with T cell ALL (T-ALL).
2. Immunophenotype
The 10%–15% of patients with T-ALL had previously been considered to have worse outcome than those with Pre-B ALL. However, survival is now similar between the 2 patient groups with appropriate treatment intensification of T-ALL patients9).
3. Genetic factors
Genetic abnormalities of the leukemic blast, including aneuploidy and recurrent translocations and deletions, are important factors in the determination of risk group and outcome in Pre-B ALL. Those that predict excellent prognosis include ETV6-RUNX1 (t(12;21)(p13;q22)) and high hyperdiploidy, that is a chromosome number ≥511011). However, even within this favorable outcome subgroup, patients with detectable minimal residual disease (MRD) at the end of remission induction may have significantly lower survival than those who are MRD(-)12). In our institution, we found that ETV6-RUNX1(+) ALL patients who were MRD(+) at the end of remission induction had EFS of 33%, in contrast to the 91% EFS for those who were MRD(-)13).
Recurrent genetic abnormalities associated with poor prognosis include BCR-ABL1 (t(9;22)(q34;q11.2)), that is Philadelphia chromosome-positive (Ph+) ALL, rearrangements of MLL (or KMT2A)(11q23), and hypodiploidy. MLL rearrangements are found in about 75% of infant ALL patients and predict a dismal outcome for this group of patients. Most patients with hypodiploid ALL have 45 chromosomes and their outcome has been reported to be similar to those with nonhypodiploid ALL14). In contrast, hypodiploid ALL patients with ≤44 chromosomes, including those with low hypodiploidy (32–39 chromosomes) and near haploidy (24–31 chromosomes), have significantly worse survival15). A recent study found that 91.2% patients with low hypodiploidy have TP53 mutations, many of whom also showed the mutations in nontumor cells, indicating that ALL patients with low hypodiploidy may have underlying Li-Fraumeni syndrome16). Hence, testing for TP53 mutations in patients with low hypodiploidy may allow for genetic counseling for those with germline mutations.
Patients with E2A-PBX1 (t(1;19)(q23;p13.3)) had previously been considered to have poor prognosis. However, patients treated on intensive, contemporary therapy were found to have favorable outcome17), and this genetic abnormality is no longer deemed a risk factor by several treatment groups. In contrast, the rare patients with E2A-HLF (t(17;19)(q22;p13.3) rearrangement have extremely poor outcome18). Intrachromosomal amplification of chromosome 21 (iAMP21) is most often diagnosed by fluorescence in situ hybridization (FISH) for the RUNX1 gene, showing five or more signals per cell in total. Patients with iAMP21 were initially deemed to have unfavorable outcome, as evidenced by the five year EFS of 29% found in one study19). However, treatment of this group of patients with high risk therapy negates the low survival associated with this genetic abnormality20).
Advances in next generation sequencing technology, including whole genome and whole exome sequencing, have aided in the identification of genetic abnormalities with prognostic relevance. One of the most critical abnormalities detected through these methods has been alteration of IKZF1, the gene for the lymphoid transcription factor IKAROS. A seminal study found IKZF1 deletion in 29% of high risk ALL patients, and confirmed that it was an independent prognostic factor for poor outcome21). Alterations of IKZF1 were frequent in patients with Ph+ ALL, as well as high risk patients with a mutational spectrum similar to that of Ph+ ALL but without the BCR-ABL1 translocation, that is Ph-like ALL. This disease subtype is known for abnormalities in kinase and cytokine receptor genes, and can be divided into those with ABL1-class rearrangements, including ABL1, ABL2, CSF1R and PDGFRB rearrangements, and those that activate JAK-STAT signaling, including JAK2, CRLF2 and EPOR rearrangements. Identification of these abnormalities is important as some of them respond to targeted therapy, with ABL1-class rearrangements responding to tyrosine kinase inhibitors (TKI) such as imatinib and dasatinib, and those with aberrant JAK-STAT signaling responding to JAK inhibitors such as ruxolitinib22). Although a simple and comprehensive method for the evaluation of these genetic abnormalities in the clinical setting is not yet readily available, some rearrangements, such as those of PDGFRB, can be detected through FISH, allowing for the identification of refractory patients who may be salvaged by TKI treatment2324).
The prognostic relevance of genetic abnormalities in T-ALL remains mostly unclear. In terms of incidence, NOTCH1 mutations are important, with over 50% of T-cell ALL patients harboring activating mutations of NOTCH125). An important subtype of T-ALL is early T-cell precursor (ETP)-ALL, characterized by an immature phenotype of CD1a (-), CD8 (-), CD5 weak, and expression of one or more myeloid or stem cell markers. The presence of FMS-like tyrosine kinase 3 (FLT3) mutations may also aid in the diagnosis of ETP-ALL. The initial study on this subtype indicated that patients with ETP-ALL had significantly worse outcome compared with other T-ALL patients26). However, ETP-ALL patients treated on UKALL 2003 had a nonsignificantly lower EFS and OS compared with other T-ALL patients27). Another study found that although ETP-ALL patients had an inferior response to initial therapy, as measured by prednisone response after one week of treatment and MRD levels, they had favorable survival, indicating that ETP status was not an independent prognostic factor when evaluated within the context of proven variables such as MRD status28). However, it is important to note that 18 of 49 patients received hematopoietic cell transplantation (HCT) in first complete remission (CR) in this study. Overall, the significance of the ETP phenotype remains unclear. However, intensive therapy including allogeneic transplant may be considered for patients who show a poor response to remission induction therapy.
4. Response to treatment
Early response to treatment may be measured by the clearance of peripheral blasts after one week of prophase steroid, which may be utilized in risk group assignment. Those with a good response to steroid, that is a peripheral blast count <1,000/mm3, have better survival compared with those with a poor response to steroid (a peripheral blast count ≥1,000/mm3)29).
MRD remains the most important prognostic factor in pediatric ALL. Current methods of measuring MRD include polymerase chain reaction (PCR) for immunoglobulin (Ig)/T-cell receptor (TCR) rearrangements, flow cytometry to detect aberrant immunophenotypes, and PCR for recurrent genetic fusions such as real-time quantitative (RQ)-PCR for BCR-ABL1. Both measurement of PCR for Ig/TCR and flow cytometry have been accurate in determining post-treatment MRD3031), with a cutoff level of >0.01% at end of induction according to flow cytometry based MRD predicting patients with significantly lower EFS12). Studies utilizing Ig/TCR PCR have also shown that MRD checked at both end of induction as well as end of consolidation is able to determine patients with significant residual disease and, hence, worse outcome3233).
Outline of treatment
1. Remission induction
Initial therapy consists of about 4 weeks of remission induction during which steroid (prednisolone or dexamethasone), vincristine, asparaginase, and intrathecal chemotherapy are given. An anthracycline, such as daunorubicin, may be administered to patients deemed high risk at diagnosis. The first intrathecal therapy, often consisting of triple therapy of methotrexate (MTX), cytarabine and hydrocortisone, is given immediately after diagnosis. This initial treatment, done to diagnose and treat central nervous system (CNS) leukemia as well as for CNS prophylaxis, is crucial to the long-term outcome of the patient as a traumatic lumbar puncture (TLP) at this step results in a greater incidence of relapse34). TLP may result in circulating leukemic blasts seeding the CNS and may also confound the diagnosis of initial CNS involvement. One study showed that delaying the initial intrathecal therapy for up to 1 week decreased the rate of TLP without adversely affecting long-term outcome35). Hence, a delay of the initial intrathecal chemotherapy for a maximum of 1 week may be considered in patients who are at high risk of experiencing TLP, such as patients with extreme hyperleukocytosis or obese adolescents with a significant number of peripheral blasts. After intrathecal administration, maintaining the patient in a prone position for one hour may increase the drug concentration in ventricular cerebrospinal fluid (CSF)36).
Asparaginase toxicity is a major impediment to chemotherapy schedule adherence, especially for adolescent patients. Aside from drug hypersensitivity, major side effects include pancreatitis, hyperglycemia, hypertriglyceridemia, coagulopathy, and thrombosis. Besides patients who show clear toxicity, drug efficacy may be compromised in asymptomatic patients due to silent inactivation37). For patients who show hypersensitivity to native Escherichia coli asparaginase, switching to the pegylated form, or a different strain such as Erwinia asparaginase, may allow for continuation of asparaginase therapy.
2. Consolidation and Intensification
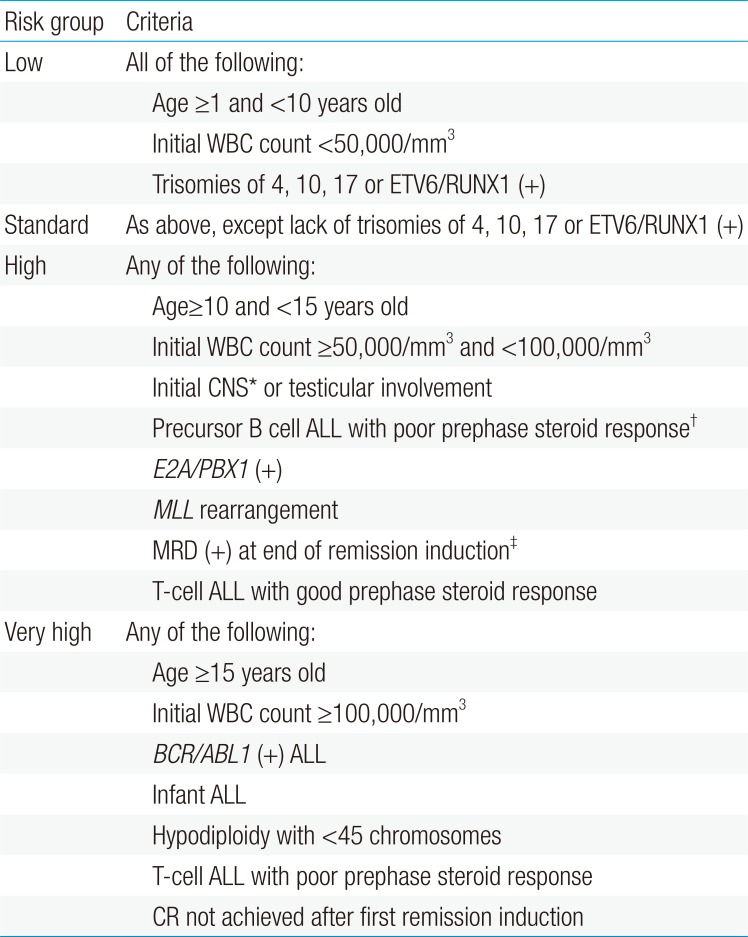
After remission induction, each patient should be classified into a risk group indicating the overall risk for relapse, based on the prognostic factors at diagnosis, and response to initial therapy, including prophase steroid response and MRD at the end of remission induction (Table 1). These risk groups predict survival, with low and standard risk groups having excellent EFS, while high risk patients have a more guarded prognosis (Fig. 1). The majority of patients will achieve CR after remission induction, subsequent to which all patients receive six months to one year of consolidation and intensification treatment, the duration and intensity of which depends on the patient risk group.

Risk group classification according to CMCP-ALL2008

The 10-year event-free survival (EFS) according to overall risk group for patients treated at our institution: low risk 91.2%±3.7%, standard risk 98.1%±1.9%, high risk 81.5%±4.3%, very high risk 59.4%±5.3%.
The consolidation phase is marked by continued CNS prophylaxis. In the past, CNS treatment was done mostly with cranial irradiation. However, the long-term neurologic and endocrine complications associated with this method of treatment have led to the omission of cranial irradiation for the majority of patients. Instead, CNS treatment for most patients consists of intensive intrathecal and systemic therapy incorporating agents such as high dose MTX38). Several studies have reported on the elimination of cranial irradiation for all patients in first CR, even for those with CNS involvement539). At our institution, we have also treated all patients in first CR without cranial irradiation regardless of initial CNS status, resulting in a cumulative incidence of any CNS relapse of 2.3%6).
Afterwards, patients receive an 8-week delayed intensification phase of treatment which utilizes drugs that were used in both remission induction and consolidation. The overall risk group of the patient is important in determining the number of intensification courses administered, with standard risk patients receiving one course of treatment to minimize toxicity40), while those with high risk features benefit from two courses of intensification41).
3. Maintenance therapy
The final phase of treatment is maintenance therapy which on average takes about 2 years for completion. The key component of this phase is antimetabolite therapy, including daily oral mercaptopurine and weekly oral MTX. Some institutions add pulses of vincristine and steroid every 4 weeks to this regimen. CNS prophylaxis should continue during maintenance therapy. As this period of treatment is prolonged and requires daily intake of medication, patient compliance is a critical issue, especially for adolescent patients. Patient noncompliance and high variability of metabolite levels within each patient, caused by varying drug doses and periods of treatment interruption, may result in an increased risk of relapse42). Patients should also continue to adhere to the Bactrim regimen of each institution, as failure to do so may result in Pneumocystis jiroveci pneumonia during this period.
4. Allogeneic HCT in first CR
The number of patients receiving HCT in first CR is decreasing, with recent studies showing 1.2%–6.6% of patients treated with upfront HCT529). An important indication for allogeneic HCT in first CR would be the small number of patients who fail to achieve CR after the first attempt at remission induction. A multinational study of 1,041 patients with induction failure showed that allogeneic HCT improved outcomes in T-ALL patients43). The strategy at our institution is to undertake HCT in first CR for patients with induction failure, BCR-ABL1 (+) ALL, hypodiploid ALL, and infant ALL with MLL rearrangement if they have a human leukocyte antigen well-matched donor.
Treatment of specific subgroups
1. Ph+ ALL
A study of a large number of patients enrolled in the UK Medical Research Council ALL97/99 trial showed a Ph+ ALL incidence of 3%11). Prior to TKI therapy, the mainstay of treatment was conventional chemotherapy combined with HCT, despite which outcome was extremely unfavorable44). The Children's Oncology Group (COG) AALL0031 trial incorporated imatinib at 340 mg/m2/day postinduction and found that continuous dosing of imatinib resulted in 5-year EFS rates comparable to those of patients who received either sibling or alternative donor bone marrow transplant45). In the EsPhALL study, based on intermittent imatinib dosing, the advantage of imatinib added to chemotherapy was less clear46). However, the study further confirmed that imatinib could safely be added to chemotherapy to improve outcomes of pediatric Ph+ ALL patients. Another study from the Spanish Cooperative Group showed that imatinib and chemotherapy followed by HCT significantly improved survival compared with historical controls47).
Despite major improvement in survival, Ph+ ALL remains a very high risk subtype of ALL with increased risk of relapse either with or without transplant. Although the results of the COG study provide evidence that HCT may not be necessary in first CR for Ph+ ALL patients, further follow-up on a larger number of patients is necessary to clarify this issue. Data from adult patients show that MRD, as measured by RQ-PCR for BCR-ABL1 during the early period of treatment, may identify patients at high risk of treatment failure48). Screening for BCR-ABL1 kinase domain mutations at relapse may identify patients who develop mutations that confer resistance to imatinib49). Finally, the follow-up period for pediatric Ph+ ALL patients treated with imatinib has been relatively short, and the long-term adverse effects of prolonged TKI treatment require further study. The deleterious impact of imatinib on bone growth, as has been observed in patients with chronic myeloid leukemia, is of particular concern for the pediatric population50).
2. Infant ALL
Infant ALL continues to have dismal outcome despite chemotherapy intensification and treatment with HCT. Cooperative studies showed long-term EFS of less than 50%751). Important prognostic factors found in these studies include the presence of MLL rearrangement, hyperleukocytosis, age less than three to six months, and poor response to prednisone prophase as having a clear adverse impact on outcome. Whether allogeneic HCT in first CR improves survival is less clear. One study showed that high risk patients with MLL rearrangements who were treated with HCT in first CR showed EFS of less than 50%52). However, data from the Interfant-99 study showed that MLL rearranged infant ALL patients had significantly better survival when treated with HCT in first CR, although patients who benefited from HCT had other poor prognosis features such as younger age, poor response to steroids, and hyperleukocytosis53). At our institution, most infant ALL patients were treated without HCT in first CR, resulting in EFS of less than 40%6). This extremely poor outcome led to a revision of treatment strategy and we currently undertake HCT in first CR for infant ALL patients with MLL rearrangement. Despite the potential benefit of HCT, infants are highly susceptible to the long-term effects of HCT on growth, development and cognition, underscoring the need for close follow-up of these parameters.
Infants without MLL rearrangement tend to be older than those with this genetic abnormality and have a more favorable outcome54). Hence, these patients may be treated with high risk chemotherapy while avoiding HCT in first CR.
3. Adolescents and young adults
Patients aged 15 and above face special issues compared with other ALL patients. Depending on the medical department to which they are transferred for care, these patients may be treated under high risk pediatric ALL protocols, or may be given chemotherapy commonly administered by adult hematologists. Although this age group may be more susceptible to the side effects of chemotherapy than younger patients, several studies have shown superior EFS compared with historical controls with acceptable toxicity when treated with pediatric regimens55). Important toxicities that are more pertinent to adolescents include steroid-related osteonecrosis, the incidence of which is significantly higher in teenagers compared with children56). Checking for medication compliance in adolescents is critical for optimal outcome, as data suggest that a significant portion of adolescent patients have difficulties in adhering to treatment schedule57).
4. ALL patients with Down syndrome
Children with Down Syndrome (DS) have a 20 fold greater risk for ALL compared with non-DS children58). Important aspects of the molecular pathogenesis of DS-ALL include CRLF2 overexpression, found in the majority of DS-ALL patients, and JAK2 mutations resulting in aberrant JAK-STAT signaling59). Outcome in DS-ALL patients is lower than in other ALL patients due to both increased rates of relapse and treatment-related mortality60). With regard to chemotherapy side effects, DS-ALL patients are more likely to develop MTX-induced toxicity, especially gastrointestinal toxicity, necessitating dose reductions of high dose MTX therapy61).
Relapsed ALL
Relapse is the most important cause of treatment failure in ALL, and a leading cause of cancer-related death in children. Long-term follow-up of a large number of relapsed patients showed survival rates ranging from 10% to over 50% depending on important prognostic factors such as site of relapse, duration of first remission, and initial risk group6263). Relapsed patients with high risk of subsequent treatment failure may be treated with allogeneic HCT in second CR, but the outcome still remains poor64).
1. Blinatumomab
As current therapeutics fail a significant portion of relapsed patients, novel treatment strategies may be the best option for this group of patients. Immunotherapy has proven to be an effective and highly promising approach for both relapsed and refractory patients, with survival rates superior to those observed with conventional chemotherapy, although longer follow-up is necessary. One of the most important agents in this regard is blinatumomab, a bispecific T-cell engager antibody that directs CD3+ effector memory T cells to CD19+ target cells including Pre-B ALL blasts65). A large study on adult ALL patients with relapsed or refractory disease showed that 43% of patients achieved CR or CR with incomplete hematologic recovery with single agent blinatumomab therapy, many of whom received subsequent allogeneic HCT66). Important adverse events in this study included neurologic events which occurred in 52% of patients, including 13% with grade 3 or above, and cytokine release syndrome (CRS). A phase I/II study was also undertaken in 70 pediatric patients with extremely poor prognosis, including refractory patients, first relapse patients unresponsive to salvage chemotherapy, and those in second or later relapse. After the first 2 cycles of treatment, 39% of patients achieved CR, half of whom were MRD negative, demonstrating that blinatumomab is an effective therapeutic option for pediatric patients with relapsed or refractory ALL67).
2. Chimeric antigen receptor (CAR) T cells
Immunotherapy utilizing autologous T cells transduced to express a receptor with specificity for CD19 has been extremely effective in relapsed/refractory ALL patients. Reported rates of CR after CAR T-cell therapy in this group of patients range from 70%–90%6869). The Children's Hospital of Philadelphia reported a 6-month EFS of 67%, with 19 out of 27 patients who achieved CR maintaining remission, most without any further therapy69). Important side effects of CAR T-cell therapy include prolonged B-cell aplasia requiring intravenous immune globulin treatment, encephalopathy and, most importantly CRS, the degree of which corresponds to leukemia burden at treatment. CRS shows laboratory findings consistent with macrophage activation syndrome, and increased levels of interleukin-6 (IL-6), allowing for treatment of this complication with the IL-6 receptor inhibitor tocilizumab. The duration of CAR T-cell efficacy most likely affects long-term EFS, with one study reporting 68% probability of CAR T cell persistence 6 months postinfusion69). An important method of CAR T cell treatment failure has been loss of CD19 expression by the leukemic blast after therapy. Such immunologic escape can be countered by the production of CAR T cells with dual targets, such as both CD19 and IL-3 receptor α-chain (CD123) as shown in a recent study70). Some of the CD19-relapsed patients have also shown the new expression of myeloid markers, or blasts consistent with acute myeloid leukemia, underscoring lineage switch as a means of evading T-cell surveillance71).
Long-term effects of treatment
With the remarkable improvement in survival of pediatric ALL compared to the past, much of the current focus is on maintaining high survival rates while limiting the long-term toxicities of treatment. Significant long-term effects include neurocognitive impairment, endocrine complications, cardiac dysfunction, and the risk for secondary malignancies72).
Neurocognitive sequelae will have a profound effect on the adult survivor's ability to function in society. The decrease in the dose of cranial irradiation currently administered compared to the past, and the elimination of cranial irradiation in many patients contribute to lessening such toxicities. One study found that patients who received 18-Gy cranial irradiation and intrathecal chemotherapy had similar neurocognitive complications to those who received intensive triple intrathecal chemotherapy with omission of cranial irradiation73). However, a study done in our cohort of survivors showed that patients who completed treatment including 18-Gy cranial irradiation performed significantly worse on standardized tests for intelligence than patients who did not receive cranial irradiation74). Regardless of the potential additional impact of CNS radiation, studies have shown that patients given other means of CNS therapy, such as multiple intrathecal infusions and systemic treatment such as high dose MTX, have also performed poorly on neurocognitive tests after treatment completion75).
Endocrine complications, such as the metabolic syndrome and short stature, may result from the use of steroids, as well as cranial irradiation. Again, the elimination of cranial irradiation in many patients may aid in minimizing these toxicities. In our study of patients treated with dexamethasone-based chemotherapy and without cranial irradiation, random glucose and body mass index decreased significantly at 12 months posttreatment completion compared to values measured during chemotherapy76). Although limited by short-term follow-up, our study did not find evidence for either long-term glucose intolerance or potential obesity during the aftermath of therapy for patients treated without allogeneic HCT and cranial irradiation. One debilitating complication of steroid therapy is osteonecrosis, with one study reporting that 60% of patients followed for 5 years since diagnosis of osteonecrosis continued to show symptoms related to this complication56).
Although patients treated on current protocols receive less of a cumulative anthracycline dose than in the past, these patients are still at risk for asymptomatic abnormalities of cardiac function that may progress to overt heart dysfunction with long-term follow-up77). Patients who have completed a treatment regimen that included anthracyclines should undergo periodic evaluation of heart function. Administering the cardioprotectant dexrazoxane along with anthracyclines may limit long-term cardiac effects78).
Conclusions
The findings from large cooperative trials have resulted in a remarkably improved survival rate for children with ALL. The key areas for future work should include studies to improve the outcome of high risk ALL, including Ph+ ALL and infant ALL, the implementation of novel therapies to treat patients who relapse, whose outcome still remains extremely poor, and the attempt to identify and minimize long-term toxicities from treatment.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.