Metformin displays in vitro and in vivo antitumor effect against osteosarcoma
Article information
Abstract
Purpose
Patients with unresectable, relapsed, or refractory osteosarcoma need a novel therapeutic agent. Metformin is a biguanide derivative used in the treatment of type II diabetes, and is recently gaining attention in cancer research.
Methods
We evaluated the effect of metformin against human osteosarcoma. Four osteosarcoma cell lines (KHOS/NP, HOS, MG-63, U-2 OS) were treated with metformin and cell proliferation was evaluated using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Cell cycle progression and apoptosis were evaluated using flow cytometric analysis, and migration and wound healing assay were performed. Fourteen female Balb/c-nude mice received KHOS/NP cell grafts in their thigh, and were allowed access to metformin containing water (2 mg/mL) ad libitum. Tumor volume was measured every 3–4 days for a period of 4 weeks.
Results
Metformin had a significant antiproliferative effect on human osteosarcoma cells. In particular, metformin inhibited the proliferation and migration of KHOS/NP cells by activation of AMP-activated protein kinase and consequent inhibition of the mammalian target of rapamycin pathway. It also inhibited the proliferation of cisplatin-resistant KHOS/NP clone cells. Analysis of KHOS/NP xenograft Balb/c-nude models indicated that metformin displayed potent in vivo antitumor effects.
Conclusion
Further studies are necessary to explore metformin's therapeutic potential and the possibilities for its use as an adjuvant agent for osteosarcoma.
Introduction
Osteosarcoma is the most common primary bone cancer, affecting children and adolescents during periods of rapid growth1). With multidisciplinary treatment, the survival of patients with localized extremity tumor is now approaching 70%1). However, the survival has reached a plateau23). Intensification of chemotherapy by adding second-line chemotherapeutic agents or dose-dense scheduling has failed to further improve the survival rate23). Patients with metastatic disease or relapsed disease still have dismal prognosis145), and new therapeutic agents based on biologic characteristics are required to improve the survival of this group of patients. Metformin is an oral hypoglycemic agent used in the treatment of type II diabetes mellitus (DM), and recently gaining attention in cancer research67). Epidemiologic studies report that metformin treatment was associated with a decreased incidence of breast, prostate, colon, and pancreatic cancers89). In retrospective studies, the use of metformin was associated with improved cancer-related mortality1011). It is suggested that metformin lowers blood glucose levels in insulin-resistant individuals, thus reducing the plasma concentration of insulin and/or insulin-like growth factor-1 (IGF1), which can act as growth factors for tumor cells12). Meanwhile, metformin exerts direct effects against cancer cells. For example, it significantly reduces the Ki67 index in primary operable breast cancer of nondiabetics13). Metformin activates AMP-activated protein kinase (AMPK), and subsequently inhibits the mammalian target of rapamycin (mTOR) pathway14). AMPK stimulates catabolic pathways to generate more energy and inhibits anabolic pathways such as protein and lipid synthesis, and mTOR stimulates energy-consuming pathways and regulates cell growth15). The features of metformin are particularly interesting for pediatric sarcomas, which have been shown to be dependent on IGF and the insulin system for their pathogenesis and progression1617). Metformin exerts antiproliferative and chemosensitizing effect both on sensitive and chemoresistant pediatric sarcoma cells16). Limited data exist about the efficacy of metformin against osteosarcoma cells 16181920). Metformin synergistically enhances antitumor activity of the histone deacetylase inhibitor trichostatin A in osteosarcoma cells, inhibits cell growth, and sensitizes osteosarcoma cells to cisplatin via cell cycle modulation18). Chen et al.20) reported that metformin inhibits metastasis and cancer stem-like sphere formation in osteosarcoma MG-63 cells. In this study, we evaluated the antitumor effects of metformin on human osteosarcoma cells. Metformin had a significant antiproliferative effect on HOS, KHOS/NP, MG-63, and U-2 OS human osteosarcoma cells. In particular, metformin inhibited the migration of KHOS/NP cells. Metformin also inhibited the proliferation of both parent and cisplatin-resistant KHOS/NP clone cells by activation of AMPK and consequent inhibition of the mTOR pathway. Finally, analysis of tumor xenograft models indicated that metformin displayed potent in vivo antitumor effects. Collectively, our data suggest that metformin deserves further investigation as an anticancer agent, implying that inhibition of both cell proliferation and metastatic potentials may significantly contribute to the treatment of human osteosarcoma.
Materials and methods
1. Cell cultures and reagents
The human osteosarcoma cell lines KHOS/NP, MG-63, and U-2 OS were obtained from the American Type Culture Collection (Manassas, VA, USA) and HOS was obtained from the Korean Cell Line Bank (Seoul, Korea). All cell lines were maintained in α-MEM medium (Life Technologies, Carlsbad, CA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Life Technologies) in a humidified atmosphere of 95% air and 5% CO2 at 37℃. Cisplatin-resistant osteosarcoma cells were derived from original parent cells and established by repeated subcultures in the presence of increasing concentrations (up to 50 µM) of cisplatin (Sigma-Aldrich, Gillingham, UK). For in vitro experiments, metformin (Selleck Chemicals, Houston, TX, USA) was dissolved in water to make a 100 mM stock solution.
2. Cell proliferation, cell cycle, apoptosis, and wound healing assay
Osteosarcoma cells (5×103/50 µL of media) were seeded in each well of 96-well plates and incubated overnight at 37℃ in a humidified 5% CO2 incubator. On the following day, 50 µL metformin was added to each well. After incubation for an additional 72 hours, cell proliferation was measured using 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide assay. The half maximal inhibitory concentration (IC50) values were determined from dose-response curves.
For flowcytometric analysis, osteosarcoma cells were plated in complete culture medium and treated with metformin (2.5 mM). Cells were harvested 24 hours later, stained with propidium iodide (1 mg/mL, Sigma, St Louis, MO, USA), and analyzed using a BD FACSCanto II Flow cytometer. Apoptosis was analyzed with an FITC Annexin V Apoptosis Detection Kit (BD Pharmingen, BD Biosciences, San Jose, CA, USA) according to the manufacturer's instructions. The fluorescent intensity was measured by BD FACSCanto Flow Cytometer (BD Biosciences).
For wound healing assay, KHOS/NP cells were seeded in 6-well plates and grown until 80% confluence. The cells were scratched with a pipette tip across the monolayer, simulating a wound. Cells were washed with phosphate-buffered saline (PBS) and cultured in α-MEM supplemented with 2% FBS, and were treated with either 1.5 mM metformin or culture medium (control). Microscopic appearance was captured 24 hours after wounding and the area of the gap region was quantified.
3. Western blotting
Cell lysates were prepared using a kit from Cell Signaling (Beverly, MA, USA). Equal amounts of protein (determined using the Bio-Rad Protein assay) were run on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and transferred to polyvinylidene difluoride membranes, which were washed with Tris-buffered saline (TBS) containing 0.1% Tween 20 (TBST), blocked with TBST and 5% wt/vol nonfat dry milk overnight at 4℃, and incubated with the following primary antibodies: CDK2, β-actin, CyclinD1 (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) and phospho-mTOR (Ser2448), phospho-mTOR (Ser2481), phospho-p70S6K (Thr389), phospho-4E BP1 (Thr37/46), phospho-AMPKα (Thr172), phospho-Acetyl-CoA carboxylase (Ser79), phospho-extracellular signal–regulated kinase (Thr202/Tyr204), phospho-AKT (Ser473) (Cell Signaling). Membranes were then washed with TBST and incubated with horseradish peroxidase-conjugated secondary antibody (1:1,000). Signals were detected using an Amersham enhanced chemiluminescence (GE Healthcare Life Sciences, Munich, Germany).
4. Tumor xenografts and metformin treatments
KHOS/NP cells (2×106) were suspended in 100-µL PBS and injected subcutaneously in the flanks of 6-week-old BALB/c nude mice (Orient Bio Inc., Seongnam, Korea). When tumors reached a volume of 50–100 mm3, mice were randomized into control and metformin-treatment groups. Metformin was dissolved in drinking water to a final concentration of 2 mg/mL. Mice were allowed to drink water, and the water was replenished every 4 days. The estimated metformin dose per mouse was 12 mg/day. Tumor volume was measured with precision calipers every 3 to 4 days for a period of 4 weeks. The tumor volume (V) was calculated using the standard formula: V (mm3)=(width)2×length/2. Experiments were conducted according to the guidelines for ethical use of animals of our Institution under an approved protocol (KIRAMS 2014-0039).
5. Statistical analysis
All data were plotted as the mean±standard error of the mean. All calculations were performed using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA, USA) and SPSS ver. 13.0 (SPSS Inc., Chicago, IL, USA), and P<0.05 was considered statistically significant.
Results
1. Effects of metformin on proliferation and migration of human osteosarcoma cells
To evaluate the antitumor effects of metformin, the in vitro antiproliferative effects of metformin (0–10 mM) were first examined against four native osteosarcoma cell lines. We found that metformin significantly inhibited the proliferation of the 4 native osteosarcoma cell lines in a dose-dependent manner and the IC50 value was 2.2–2.9 mM (Fig. 1A). To simulate the clinical setting of recurrent or refractory osteosarcoma, we established cisplatin-resistant cells by exposing KHOS/NP and U-2 OS cells to cisplatin (Fig. 2A) and investigated the effect of metformin on cell proliferation. Metformin inhibited the proliferation of both native and cisplatin-resistant KHOS/NP and U-2 OS cells. The IC50 values of metformin were higher in cisplatin-resistant cells than in native cells, 4.2–4.5 mM vs. 2.2–2.9 mM, respectively (Fig. 2B). Therefore, we concluded that metformin inhibited the proliferation of both native and cisplatin-resistant osteosarcoma cells. Since metformin has been demonstrated to have antimetastatic effects, we examined the effect of metformin on the migration of osteosarcoma cells. We found that metformin treatment significantly inhibited the migration of KHOS/NP cells via analysis of wound-healing (Fig. 1B). From these results, we suggest that metformin could potentially inhibit proliferation and migration of human osteosarcoma cells. Although metformin inhibited cell proliferation, it did not induce apoptosis of KHOS/NP cells (Fig. 1C). To determine the effect on cell cycle, KHOS/NP cells were treated with 2.5 mM metformin for 24 hours, stained with propidium iodide, and analyzed using flow cytometry. As shown in Fig. 3A, compared with control, the fraction of G0/G1 cells increased after metformin treatment (from 43% to 51.3%). Moreover, we found that the levels of cyclin D1 and CDK2, key regulators for the G1/S transition, were decreased (Fig. 3B). This result indicated that metformin induced cell cycle arrest at IC50 (2.5 mM) in KHOS/NP cells.
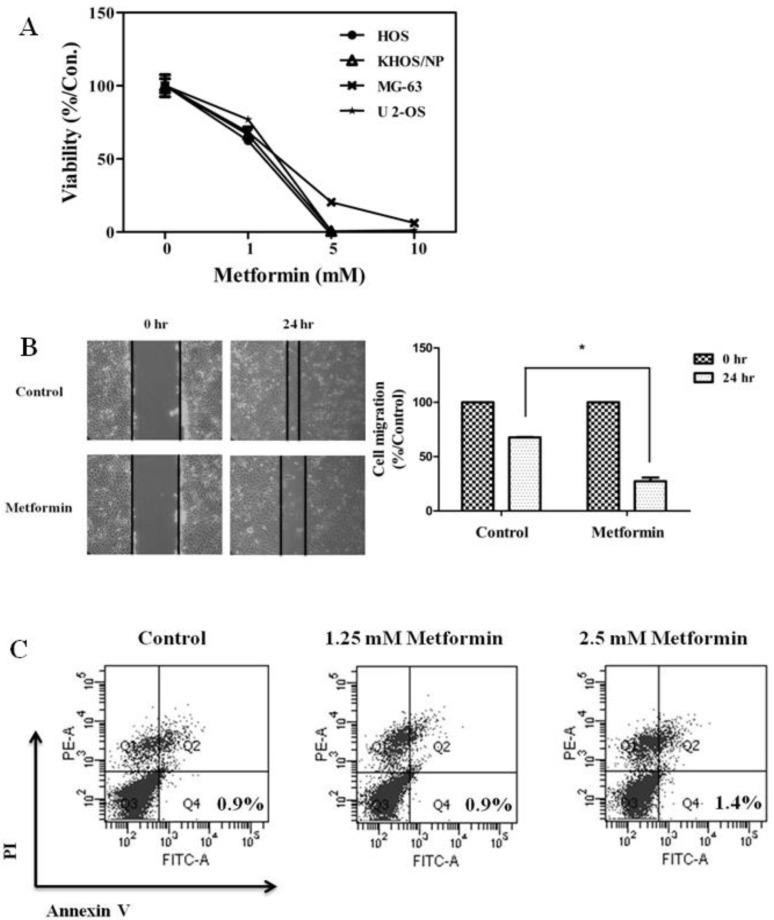
(A) Metformin decreased the proliferation and migration of osteosarcoma cells. Four osteosarcoma cells were treated with metformin (0–10 mM) and cell viability assay (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) was performed 72 hours later. (B) KHOS/NP cells were plated in 6-well plates and grown to 80% confluence. After scratching, cells were incubated with or without metformin (1.5 mM). Microscopic appearance was captured 24 hours after wounding. The area of the gap region was quantified by ImageJ and data are shown in the bar graph (*P<0.05). (C) KHOS/NP cells were treated with metformin (1.25 or 2.5 mM) for 24 hours. Apoptotic cells were detected by Annexin V-propidium iodide (PI) dual staining.

Metformin decreased the proliferation of cisplatin-resistant osteosarcoma cells. Cisplatin-resistant osteosarcoma cells were established (A) and treated with metformin (B) and cell viability assay was performed 72 hours later. The half-maximal inhibitory concentration (IC50) values were determined from dose-response curves.
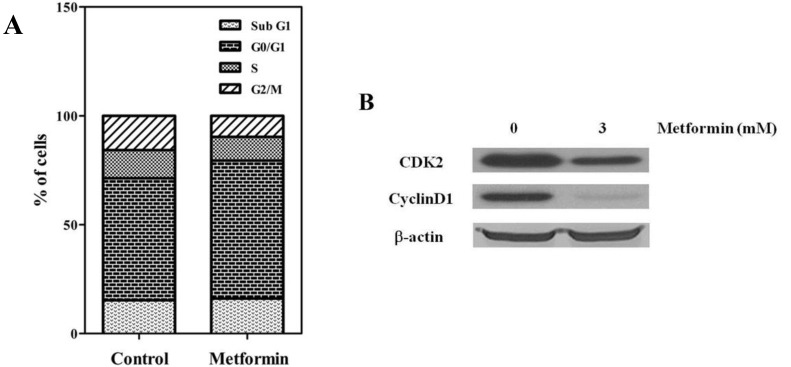
Metformin induced cell-cycle arrest of KHOS/NP cells. KHOS/NP cells were treated with 2.5–3 mM metformin for 24 hours. (A) Cell cycle was analyzed by flow cytometry; (B) the CDK2 and cyclin D1 protein expression of KHOS/NP cells after metformin treatment.
2. Metformin activates the AMPK pathway and inhibits mTOR and its downstream pathways
Metformin activity has been responsible of activation of AMPK and its inhibitory function on mTOR. To test the functional activation of AMPK by treatment with metformin, we examined the phosphorylation level of both AMPK and acetyl CoA carboxylase, a target of AMPK known to be one of the substrates of AMPK activity, in KHOS/NP cells. Twenty-four hours after treatment with 3 mM metformin, p-AMPK (Thr172) and p-ACC (Ser79) were visible in KHOS/NP cells (Fig. 4A, B). To further elucidate the underlying mechanism, levels of mTOR and its substrates were measured. We found that metformin inhibited the phosphorylation of mTOR (Ser2481), mTOR (Ser2448), P70S6K, 4E-BP1, and ERK (an indicator for mTOR activity) (Fig. 4C).
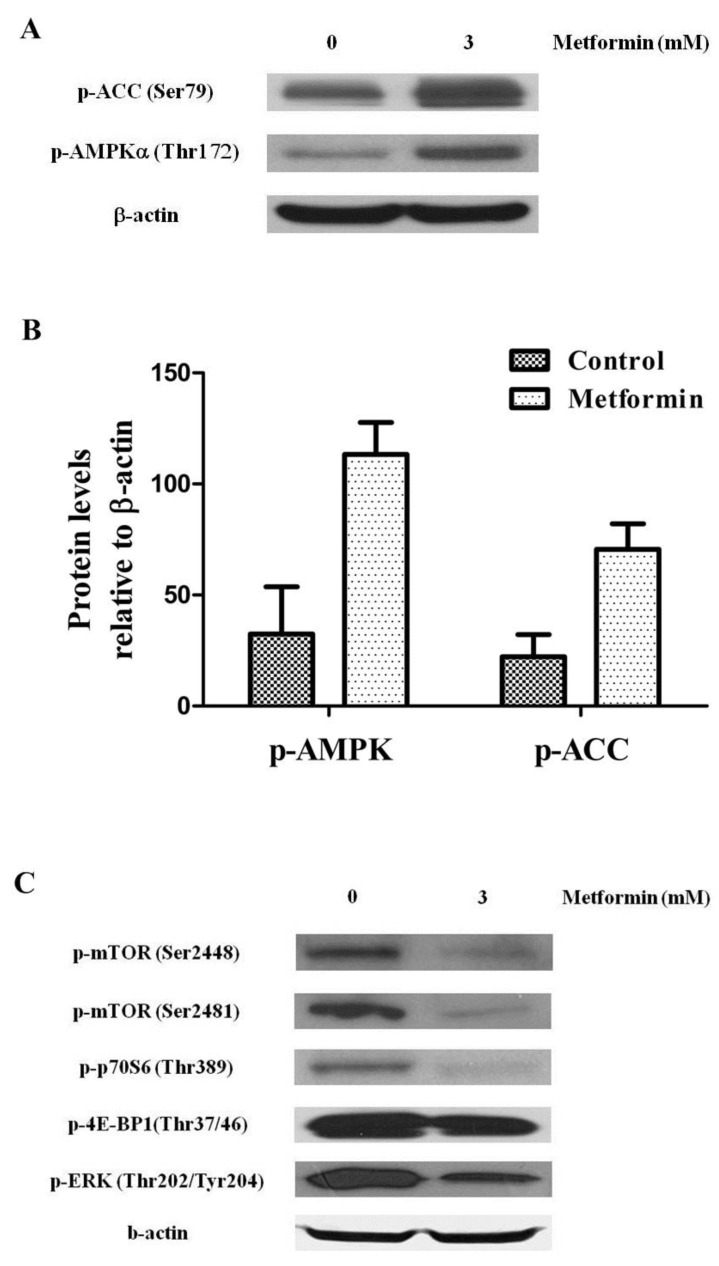
Metformin controlled the AMP-activated protein kinase (AMPK)/mammalian target of rapamycin (mTOR) signaling pathway. KHOS/NP cells were treated with 3 mM metformin for 24 hours and Western blot analysis was performed to examine the levels of phospho-AMPK (p-AMPK) (T172), phospho-Acetyl-CoA carboxylase (p-ACC) (S79) (indicator for AMPK activity) (A, B) and mTOR and its substrates (C) in KHOS/NP cells.
3. Metformin inhibits the growth of KHOS/NP tumor xenograft
To determine whether metformin could curtail tumor growth in vivo, BALB/c nude mice were injected with KHOS/NP cells and treated with metformin (approximately 12 mg/day/mouse for 4 weeks). Metformin significantly inhibited the growth of KHOS/NP tumors in BALB/c nude mice (Fig. 5). From the 14th day of study entry, tumor volume of the metformin-treated mice was significantly smaller than that of the control mice (P<0.05). When the mean tumor volume was compared using fold increase at the 27th day, fold increase of the metformin-treated mice was 48% that of the control mice.
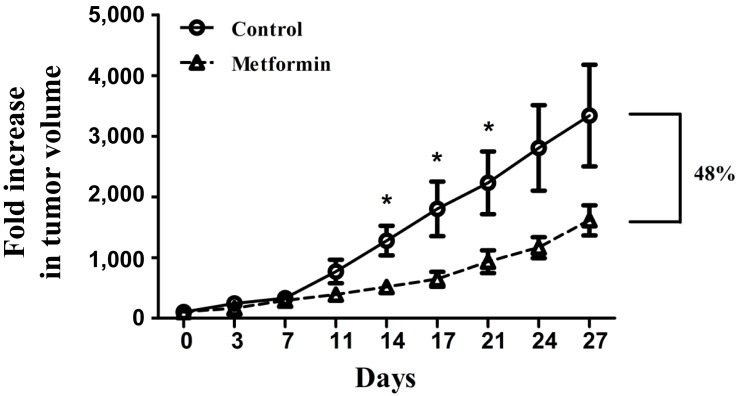
Metformin inhibited the growth of KHOS/NP xenograft tumor of Balb/c-nude mice. Graphs represent mean±standard error of the mean of five tumors per condition (*P<0.05).
Discussion
In the present study, we have observed the in vitro and in vivo effects of metformin against osteosarcoma. Metformin decreased the proliferation and migration of osteosarcoma cells, and inhibited the growth of KHOS/NP osteosarcoma in a tumor xenograft model. In addition, metformin showed antiproliferative effects on cisplatin-resistant osteosarcoma cells, which were established as an in vitro model of recurrent or refractory osteosarcomas. Our findings suggest the possibility of metformin being used as an adjuvant agent in the treatment of recurrent or refractory osteosarcoma patients.
As in previous reports, we observed that metformin activated the AMPK pathway and downregulated the mTOR pathway of osteosarcoma cells. Recent genomic study suggested the PI3K/mTOR pathway as a common vulnerability in osteosarcoma21), and we assume that our findings suggest the constitutive over-activation of the PI3K-mTOR pathway in osteosarcoma cells. It is reported that metformin inhibits the migration and epidermal-mesenchymal transition of thyroid cancer cells22). Similarly, we observed that metformin decreased the migration of osteosarcoma cells. The critical factors regulating the growth, migration, and invasion of tumor cells are cyclin D1 and c-Myc 192223). Although it is presumed that metformin inhibits these factors through AMPK activation, another mechanism suggested to be involved in this phenomenon is the Akt-MDM2-Foxo3a signaling axis24). Our observation, together with other reports, indicated that either the AMPK-dependent or independent mechanism of metformin might be implicated in inhibition of proliferation and migration of human osteosarcoma.
Controversy exists about the association between the effect of metformint and TP53 or RB mutation status. We observed that metformin induces cell cycle arrest of both KHOS/NP (mutated TP53 and wild type RB) and U-2 OS cells (wild type TP53 and RB, data not shown). For U-2 OS cells, metformin (2.5 mM) treatment increased the expression of TP53 and RB protein (data not shown). For KHOS/NP cells, metformin treatment did not induce any changes of RB and TP53 protein expression, but decreased the expression of cyclin D1 protein (Fig. 3B). It is reported that metformin treatment phosphorylates the Chk1 of TP53 wild-type osteosarcoma cells, but not that of TP53 null MG-63 cells19). AMPK activation induces a p53-dependent metabolic G1/S checkpoint in response to low glucose conditions25). On the contrary, other studies suggest that TP53 loss increased the growth inhibitory and toxic effect of metformin on cancer cells26). In the present study, effects of metformin did not appear to be influenced by TP53 mutation status of osteosarcoma cells.
The optimal dose of metformin required to elicit antitumor effect remains elusive. The in vitro concentration of metformin showing antitumor effect was in the millimolar range, as the IC50 value 3 mM of our study141618). However, the serum levels of metformin achieved in DM patients and in many in vivo models were in the micromolar ranges27). The epidemiologic data suggest that usual recommended doses of metformin might have antitumor effect89). It is uncertain what metformin concentrations are achieved in tumor tissues of patients receiving conventional antidiabetic doses of metformin. We assume that the high concentrations of glucose and growth factors in cell culture media, along with the presence of oncogenic mutations and unknown expression of the membrane transporter OCT1, which is essential for the transport of metformin across cell membrane28), may account for the higher concentrations of metformin required to elicit in vitro antitumor effect29).
At present, clinical trials are underway to evaluate the effect of metformin as a single agent, or in combination with cytotoxic chemotherapy, and/or radiotherapy for the treatment of various cancers30). The preliminary results from patients with breast and endometrial cancer have shown that metformin can significantly impact markers of tumor proliferation313233). Currently ongoing clinical trials are designed to assess the survival benefit of metformin in patients with breast, pancreas, lung, endometrial, brain, prostate, and gynecological cancers30).
Summarizing, we observed that metformin has some efficacy against osteosarcoma and suggest the possibility of using it as an adjuvant agent in the treatment of osteosarcoma. Our findings are based on preclinical data and a large-scale prospective study is required to validate our preclinical findings.
Acknowledgment
This study was supported by a grant from the Korea Institute of Radiological and Medical Sciences (KIRAMS), funded by the Ministry of Science, ICT and Future Planning, Korea (1711021931).
Notes
Conflicts of interest: No potential conflict of interest relevant to this article was reported.