Pathogenesis of minimal change nephrotic syndrome: an immunological concept
Article information
Abstract
Idiopathic nephrotic syndrome (INS) in children is characterized by massive proteinuria and hypoalbuminemia. Minimal change nephrotic syndrome (MCNS) is the most common form of INS in children. The pathogenesis of MCNS still remains unclear, however, several hypotheses have been recently proposed. For several decades, MCNS has been considered a T-cell disorder, which causes the impairment of the glomerular filtration barrier with the release of different circulating factors. Increased levels of several cytokines are also suggested. Recently, a "two-hit" theory was proposed that included the induction of CD80 (B7-1) and regulatory T-cell (Treg) dysfunction, with or without impaired autoregulatory functions of the podocyte. In contrast to the well-established involvement of T cells, the role of B cells has not been clearly identified. However, B-cell biology has recently gained more attention, because rituximab (a monoclonal antibody directed against CD20-bearing cells) demonstrated a very good therapeutic response in the treatment of childhood and adult MCNS. Here, we discuss recent insights into the pathogenesis of MCNS in children.
Introduction
Idiopathic nephrotic syndrome (INS) is a common chronic illness characterized by massive proteinuria and hypoalbuminemia in children12). Massive urinary loss of serum proteins can cause a hypercoagulable state, the dysregulation of fluid, electrolyte imbalance, and susceptibility to infections3). The International Study of Kidney Disease in Childhood reported that 84.5% of children with INS had minimal change nephrotic syndrome (MCNS), 9.5% had focal segmental glomerulosclerosis (FSGS), 2.5% had mesangial proliferative glomerulonephritis, and 3.5% had membranous nephropathy or other diseases leading to nephrotic-range proteinuria4). The pathological hallmark of MCNS is the effacement of foot processes in glomeruli, revealed by ultrastructural analysis, without any inflammatory injury or immune complex deposition2).
Eighty to ninety percent of children with INS respond to steroid treatment2). Unfortunately, 60%–80% of the children with steroid-responsive nephritic syndrome relapse, but they almost never develop end-stage renal disease (ESRD)2). Resistance to immunosuppressant therapy, including glucocorticoid and cyclosporin A, occurs in 50% of FSGS cases and 10% of MCNS cases, which is associated with progression to ESRD.
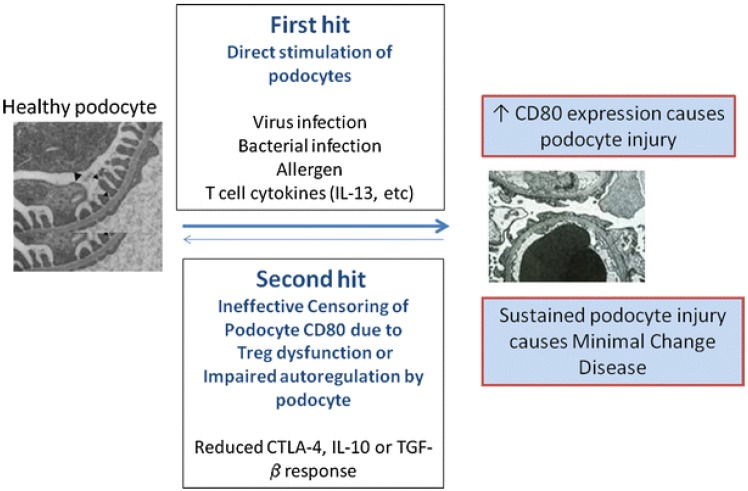
More than four decades ago, MCNS was considered as an exclusive systemic disorder of T cells and cell-mediated immunity5). Increased levels of various cytokines were also suggested as one aspect of the pathogenesis. Recently, Shimada et al.6) proposed a "two-hit" theory that included the induction of CD80 (or B7-1) and regulatory T-cell (Treg) dysfunction, with or without impaired autoregulatory function of the podocytes (Fig. 1). B-cell biology has also attained great attention, since rituximab, a monoclonal antibody directed against CD20-bearing cells, showed good therapeutic potential in the treatment of both childhood and adulthood MCNS789). However, the precise pathophysiology of MCNS still remains elusive. We hereby discuss immunological insights and recent findings on the pathogenesis of MCNS.
T-cell signal transduction and abnormal Tregs
Approximately 40 years ago, Shalhoub5) proposed a major hypothesis that MCNS was caused by a circulating factor derived from dysfunctional T cells. This hypothesis was based on several findings, such as the presence of no immune complexes in glomeruli, a good response to steroids, and frequent remission after measles infection, which led to cell-mediated immuno-suppression5). For example, treatments of T-cell suppressive drugs, such as cyclosporin A and basiliximab (an anti-interleukin [IL]-2 receptor antibody), were effective in some patients with MCNS210).
In the last decades, it has been found that Tregs are a distinct subset of T-lymphocytes that play a pivotal role in maintaining immune homeostasis and tolerance to self-antigens11). The dysregulation of Tregs has been shown to be important in the pathogenesis of several autoimmune diseases, such as rheumatoid arthritis and systemic lupus erythematosus12). Recently, it has also been suggested that Tregs play an important role in the pathogenesis of MCNS1314). Shimada et al.6) reported Treg dysfunction and/or impaired autoregulatory function by the podocyte have the potential to turn off CD80 expression once it is induced. Treg dysfunction could make transient massive proteinuria persistent, leading to podocyte injury, and eventually, MCNS6). Transient massive proteinuria is typically triggered by viral infections, bacterial infections, or allergen- or T-cell-mediated release of cytokines (e.g., IL-13, etc.)6).
The importance of the association between Tregs and nephrotic syndrome is highlighted by immune dysregulation, polyendocrinopathy, enteropathy, and X-linked (IPEX) syndrome with concomitant nephrotic syndrome15). IPEX syndrome is a rare disorder of the immune regulatory system caused by mutations of FOXP3, which is a transcription factor responsible for the generation and maturation of Tregs, and the development of MCNS in IPEX syndrome can be explained by Treg dysfunction15). However, the reason behind children with MCNS having Treg impairment and/or podocyte autoregulation is still unknown.
Cytokine profiles in MCNS
Some studies have shown an association between various cytokines and proteinuria, and stated that glomerular permeability factors can be responsible for nephrotic syndrome in patients or in animal models of the proteinuric disease16). Accordingly, the increase in monocyte/macrophage cytokines, including IL-1, IL-12, and tumor necrosis factor-alpha (TNF-α), was important in the initiation and recurrence of INS17). In rats with adriamycin-induced nephrotic syndrome, IL-1 produced by resident glomerular macrophages was associated with proteinuria18). In supernatants from children with MCNS, the levels of IL-1 were found to be elevated19).
IL-12, secreted by dendritic cells and macrophages, is also recognized as a T cell-stimulating factor20). It enhances the cytotoxic activity of natural killer cells and cytotoxic T-lymphocytes21). IL-12 levels were increased during the active clinical phase of MCNS in peripheral blood monocyte cultures (PBMC)22). On the contrary, some reports could not detect any IL-12 in serum, urine, and supernatants of PBMC from children with steroid sensitive nephritic syndrome (SSNS)23). Serum IL-12 levels were not significantly disparate between the active phase and remission phase of SSNS24).
TNF-α was increased in the blood and urine of children with NS, and mRNA expression was also increased in PBMC of these patients2526). TNF-α has been found to induce glomerular injury in experimental MCNS25). IL-15 derived from mononuclear phagocytes following infections induced the differentiation of immature T-helper cells27). This cytokine mimics the stimulatory function of IL-2 on T cells and is believed to cause the release of the vascular permeability factor by PBMCs from nephrotic patients in combination with IL-1228).
In addition to these cytokines, other monocyte/macrophage-related cytokines are IL-6, IL-8, IL-10, interferon-gamma (IFN-γ), and IL-18293031323334353637383940). IL-6 expression in the urine and renal tissues was correlated with proteinuria in MCNS rats9). Also, serum IL-8 levels were higher in the initial nephrotic phase than in the remission phase in children with SSNS, and urinary concentrations of IL-8 were correlated with proteinuria in children with NS3233). In contrast, however, Daniel et al.34) reported that serum IL-8 levels were decreased in cases of SSNS compared to healthy controls. IL-10 is mainly produced by monocytes, which have the capacity to reduce the release of proinflammatory cytokines, such as IFN-γ, IL-2, and TNF-α35). In the study by Matsumoto36), the release of IL-10 was lower in the supernatants of PBMC from patients with MCNS than from that of controls.
T-helper cells, cytotoxic T cells, natural killer cells, and macrophages secrete IFN-γ, which is shown to promote Th1 differentiation, eventually leading to cellular immunity and simultaneously inhibiting Th2 differentiation37). The concentration of IFN-γ was not increased in cases of relapse of children with SSNS3839). Serum IFN-γ was significantly lower in the active phase of NS compared with the remission phase40).
IL-18 is produced by macrophages, and in combination with IL-12, it plays a pivotal role in cell-mediated immunity following infection41). Urinary levels of IL-18 correlated with disease activity in patients with MCNS42). Similarly, IL-18 levels in vitro were related to the disease activity in MCNS patients43). Serum levels of IL-18 correlated with both IL-4 and IL-13 in childhood SSNS40).
IL-2 promotes the differentiation of immature T cells into Tregs and is implicated in the "battle" against infections and prevention of autoimmune diseases44). IL-2 concentrations were significantly increased during relapse when than that during remission in children with SSNS343845). The IL-2 mRNA expression was also significantly higher in the acute phase than in the remission phase of childhood INS46).
Th2 cytokines, such as IL-13, have been highlighted in the pathogenesis of MCNS39474849). CD4+ and CD8+ IL-13 mRNA expression increased during relapse than that during remission in children with SSNS3947). IL-13 overexpression led to podocyte injury in MCNS rat models48). Serum IgE levels are elevated during relapse in SSNS and were correlated with IL-13 upregulation47).
Overexpression of IL-13 and CD 80 (B7-1) in MCNS
Recent studies have identified that increased IL-13 expression can lead to podocyte injury and can induce a MCNS-like phenotype395750). An increase in IL-13 production by CD3+, CD4+, and CD8+ T cells was shown to mediate SSNS3947). Although no significant histologic changes were observed in the glomeruli of IL-13-transfected rats, they clinically exhibited substantial proteinuria, hypoalbuminemia, and hypercholesterolemia48). Electron microscopy revealed up to 80% effacement of podocyte foot processes, which progressed to nephrotic syndrome48). Notably, overexpression of IL-13 caused the downregulation of nephrin, podocin, and dystroglycan. These proteins are critical molecules in maintaining slit diaphragm (SD) integrity, and the concurrent upregulation of CD80, IL-4Rα, and IL-13Rα2 in IL-13-transfected rats48).
More recently, Park et al.5152) reported that IL-13 significantly decreased zonula occludens-1 (ZO-1) protein levels in human podocytes, whereas ZO-1 protein production was significantly increased in a rat model of puromycin aminonucleoside nephrosis. They demonstrated that IL-13 alters the expression of ZO-1, and such alterations in the content and distribution of ZO-1 may also be relevant to the pathogenesis of proteinuria in the MCNS model, which was significantly restored after treatment with a leukotriene receptor antagonist51). Therefore, these findings can further strengthen the hypothesis that IL-13 may increase podocyte permeability through the modulation of SD proteins, resulting in nephrotic-range proteinuria, namely MCNS, and also provide an explanation for the plausible connection among Th2 cytokines, MCNS, and atopy.
Increased IL-13 also induced the upregulation of CD80, IL-4Rα, and IL-13Rα2. CD80, a cell surface glycoprotein expressed on activated B lymphocytes, is a dendritic-associated receptor that acts as a costimulatory signaling molecule of the T cell when bound to CD28 expressed on T cells53). Exposure to low-dose lipopolysaccharide (LPS) was shown to upregulate CD80 in the podocytes of wild type and severe combined immunodeficient mice in vivo, which caused nephrotic-range proteinuria53). Mice lacking CD80 are protected from LPS-induced NS, which suggests a link between podocyte CD80 expression and proteinuria53). Tregs are known to inhibit the immune response by releasing soluble cytotoxic T-lymphocyte antigen 4 (sCTLA-4), IL-10, and transforming growth factor (TGF)-β, which can suppress CD80 expression on the antigen presenting cells (APCs). This leads to a blockade of the costimulatory activation of T cells54). Therefore, the immune response is initiated by the activation of CD80 on APCs, and over time, negatively regulated, with the Treg playing a pivotal role in this process.
In MCNS, it has been supposed that local mediators, such as podocyte-derived angiopoietin-like-4 (Angptl-4) and CD80, may be implicated in the pathogenesis of MCNS55). Theoretically, a number of cells within the human kidney may be able to secrete CD80, including podocytes, macrophages, dendritic cells, and tubular cells54). Overexpression of CD80 in podocytes has been observed in genetic, immune-mediated, drug-induced, and bacterial toxin-induced experimental kidney diseases with nephrotic syndrome56).
Urinary soluble CD80 was significantly increased in MCNS patients during relapse when compared with healthy controls and MCNS patients during remission or with FSGS56). Urinary CD80 correlated with disease activity. The urinary CD80/CTLA-4 ratio was more than 100 folds higher in patients with relapsed MCNS compared with those in remission. In addition, CD80 was observed in glomeruli by immunohistochemical staining in seven biopsy specimens of eight patients with MCNS during relapse56). Western blotting was also performed to distinguish between urinary soluble CD80 (MW 23 kDa) and membrane-associated CD80 (MW 53 kDa). Urinary CD80 was present as an approximately 53 kDa large protein, which suggests cell membrane-associated urinary CD8056). CD80 expression and CD80 protein secretion by podocytes were significantly increased in sera from patients with MCNS in relapse compared with sera from patients in remission53). Further studies addressed if CD80 and soluble urokinase plasminogen activator receptor (suPAR) levels are able to be reliably used to distinguish between MCNS and FSGS. However, urinary suPAR levels were not distinguishable between both diseases, while urinary CD80 levels were significantly increased in MCNS patients in relapse compared to those in remission and FSGS patients55).
LPS was capable of enhancing the expression of CD80 in podocytes, leading to nephrotic-range proteinuria53). CD80 was colocalized with the podocyte synaptopodin in human and murine kidney tissue specimens53). Activation of CD80 in cultured podocytes led to the reorganization of vital SD proteins, whereas LPS-signaling pathway through the toll-like receptor (TLR)-4 led to the reorganization of the podocyte actin cytoskeleton in vivo53). Therefore, CD80 may contribute to the pathogenesis of proteinuria by disrupting the SD structure53).
To depict pathogenetic pathways regulating and driving CD80 induction in podocytes, several studies were performed5758). Polyinosinic-polycytidylic acid (polyIC) induced types I and II interferon signaling, nuclear factor kappa B activation, and the induction of CD80 expression, but dexamethasone blocked both basal and polyIC-stimulated CD80 expression, as did the inhibition of nuclear factor kappa B57). Intravenous injection of polyIC-LMW into mice resulted in significant albuminuria and led to increased urinary CD80 excretion58). Partial foot process fusion in glomeruli was seen by electron microscopy58).
It has been proposed that MCNS is a "two-hit" disorder6). The initial hit is the induction of CD80 on the podocyte, resulting in an alteration in shape with actin disruption that causes increased glomerular permeability and proteinuria6). The second hit is usually caused by the ineffective censoring of podocyte CD80 due to Treg dysfunction or impaired podocyte autoregulation and reduced CTLA-4, IL-10, or TGF-β response. CD80 expression may result from either direct binding of the podocyte by activated T-cell cytokines such as IL-13, or by activation of podocyte TLR by microbial products or allergens6). In normal circumstances, CD80 expression on podocytes is terminated by Treg cytokines or CTLA-4 and IL-10 by podocytes. However, if a second hit occurs in MCNS and abnormal censoring of podocyte CD80 expression continues due to a defective autoregulatory response by Tregs or by the podocyte itself, CD80 expression persists and results in MCNS6). An important experiment that has yet to be done is that of podocyte specific overexpression of CD80, which would clarify a direct effector role of CD80 in inducing proteinuria.
What do we know about the role of circulatory factors in MCNS?
The implicated role of circulatory factors in the etiopathogenesis of MCNS has long been postulated. Vascular permeability factor, elaborated from concanavalin A-stimulated lymphocytes obtained from patients with MCNS, acts on systemic capillaries and on the glomerular permeability barrier59). The effects on the vasculature mimic the effects of IL-2 on permeability59). However, lymphocytes from MCNS patients with high vascular permeability factor activity revealed low amounts of IL-2, and immunoadsorption leading to the complete removal of IL-2 did not affect vascular permeability factor activity60). It became apparent that its secretion is increased by IL-2, IL-12, IL-15, and IL-18, whereas the addition of TGF-β to concanavalin A-stimulated MCNS T cells inhibited the release of the vascular permeability factor61).
In 1999, it was hypothesized that the vascular permeability factor 100KF, which has a role in MCNS, is closely related to hemopexin62). Plasma hemopexin was capable of inducing significant proteinuria and glomerular alterations, resembling MCNS63). Furthermore, it was shown that the mean titer of hemopexin is decreased during relapse, and compared to remission samples and other glomerulopathies, significant upregulation of hemopexin plasma activity and different hemopexin fragments were demonstrated during relapse64). It was concluded that active hemopexin could be present in an altered isoform64). Hemopexin-treated human podocytes showed marked actin reorganization after 30 minutes, which was reversible after four hours and led to the activation of protein kinase B, as well as RhoA. Moreover, Lennon et al.65) postulated a nephrin-dependent process, since these changes did not occur in nephrin-deficient podocytes.
Interestingly, a role for the glomerular secretion of Angptl-4 was proposed66). A podocyte-specific transgenic model (NPHS-Angptl-4) revealed a 500-fold increase in albuminuria in homozygous males over time, whereas adipose tissue-secreted Angptl-4 could not induce any proteinuria66). Electron microscopy showed a selective Angptl-4 signal in glomeruli and foot-process effacement in 5-month-old homozygous rats66). Glucocorticoid treatment reversed the sharp increase of Angptl-4 mRNA expression66). The initial enthusiasm was hampered due to the finding that Angptl-4 is increased in other diseases, leading to nephrotic-range proteinuria as well67). Angptl-4 is secreted in response to an elevation in the ratio of plasma free fatty acids to albumin, in terms of heavy proteinuria, reducing proteinuria but resulting in hypertriglyceridemia67).
B-cell abnormality and efficacy of rituximab
In contrast to the well-established involvement of T cells in MCNS, the role of B cells is not yet certain. Despite an observed association between allergic diathesis and the onset of MCNS, an elevation of IgM, IgE, B-lymphocytes, and their subsets, surface IgM-, IgG-, and IgE-positive cells (Bγ, Bµ, and Bɛ) were already described in an earlier study68). In nephrotic patients, an increase in cytoplasmic Bγ (cBγ) was shown with an elevated cBγ/surface Bγ, which normalized during steroid treatment and increased again after withdrawal69). Recently, it was also shown that nuclear factor-related kappa B is upregulated during the relapse of MCNS, mainly in CD4+ T cells and B cells, and this induces the activation of AP1 signaling70).
B-cell biology, however, has attained more attention lately, since treatment with rituximab, a monoclonal antibody directed against CD20 bearing cells, has shown good therapeutic responses in the treatment of both childhood and adulthood MCNS7717273). Children with frequently relapsing nephrotic syndrome or steroid-dependent nephrotic syndrome had a significantly longer relapse-free period in the rituximab group compared to the control group7873). Moreover, fewer patients with a significantly longer time to treatment failure were reported in the rituximab group7). Concurrent steroid treatment could be significantly reduced following rituximab initiation, while the rate of serious adverse events did not differ between both groups7). Subsequently, rituximab treatment has been licensed for the treatment of steroid-dependent and frequently relapsing nephrotic syndrome by the Ministry of Health, Labor and Welfare in Japan74).
Conclusions
Here, we reviewed the pathogenesis of MCNS from an immunological perspective. Recent studies propose that the pathogenesis of MCNS could involve both lymphocytes and podocytes. Further studies are required to elucidate the exact pathophysiology of MCNS, and the development of novel drugs that target podocytes and immunosuppressants for lymphocytes are also needed.
Acknowledgments
Andreas Kronbichler was supported by the ERA-EDTA with a long-term fellowship (12 months) from August 2014 to August 2015. Jae Il Shin was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) and funded by the Ministry of Education, Science and Technology (2011-0013789, 2013R1A1A1012112 and 2015R1C 1A1A01052984).
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.