Kabuki syndrome: clinical and molecular characteristics
Article information
Abstract
Kabuki syndrome (KS) is a rare syndrome characterized by multiple congenital anomalies and mental retardation. Other characteristics include a peculiar facial gestalt, short stature, skeletal and visceral abnormalities, cardiac anomalies, and immunological defects. Whole exome sequencing has uncovered the genetic basis of KS. Prior to 2013, there was no molecular genetic information about KS in Korean patients. More recently, direct Sanger sequencing and exome sequencing revealed KMT2D variants in 11 Korean patients and a KDM6A variant in one Korean patient. The high detection rate of KMT2D and KDM6A mutations (92.3%) is expected owing to the strict criteria used to establish a clinical diagnosis. Increased awareness and understanding of KS among clinicians is important for diagnosis and management of KS and for primary care of KS patients. Because mutation detection rates rely on the accuracy of the clinical diagnosis and the inclusion or exclusion of atypical cases, recognition of KS will facilitate the identification of novel mutations. A brief review of KS is provided, highlighting the clinical and genetic characteristics of patients with KS.
Introduction
Kabuki syndrome (KS, OMIM#147920) is a rare syndrome characterized by distinct dysmorphic facial features, postnatal growth retardation, intellectual disabilities, skeletal abnormalities, and unusual dermatoglyphic patterns. It is usually detected sporadically and has a wide spectrum of clinical manifestations1). Recently identified as a genetic syndrome via whole exome sequencing2), KS has been increasingly recognized in the primary care setting, and in many cases, its underlying etiology has been discovered. KS is diagnosed and managed by pediatricians. To enhance the understanding of patients with KS among pediatricians, this review focuses on the clinical and molecular genetic characteristics of these patients.
What is KS?
In 1969, Japanese geneticist Norio Niikawa encountered a young patient presenting with unusual facial features and mental retardation. Whether this was a never before diagnosed condition with a genetic underpinning was unclear. Over the next several years, Niikawa treated several other children with the same symptoms in his outpatient clinic, furthering support for a new disorder1). In 1981, two independent groups in Japan separately described this disorder in the Journal of Pediatrics3). Because its characteristic facial features resembled the make-up of actors in Kabuki, the traditional Japanese theater, the term 'Kabuki make-up syndrome' was coined. The most remarkable aspects of KS are the atypical facial features, which usually prompt the clinician to consider this diagnosis4). Patients often have long palpebral fissures with eversion of the lateral one-third of the lower eyelid, arched eyebrows with sparseness in the lateral one-third, short columella with a depressed nasal tip, prominent ears, other congenital anomalies, and various health problems4).
How common is KS?
KS was initially thought to be specific to Japanese individuals. Its estimated prevalence in Japan is 1/32,0005). However, there are now reports of KS in a variety of ethnic groups, including Northern European, Brazilian, Vietnamese, Filipino, East Indian, Arabic, Chinese, Mexican, and African4). The global incidence of KS is not known. Although KS is reportedly rare in Korea, its prevalence in this country may be underestimated because of under-recognition of the diagnosis and the paucity of diagnostic tools. Considering the many reports of other populations, the frequency of KS in the Korean population presumably approximates that of the Japanese population. Recently, a multicenter study6) confirmed that number of patients newly diagnosed with KS has increased owing to high-quality comprehensive diagnostic services beginning in 2000. A population-based study is required to accurately determine whether the incidence of KS is rising in Korea.
How can we clinically diagnose KS?
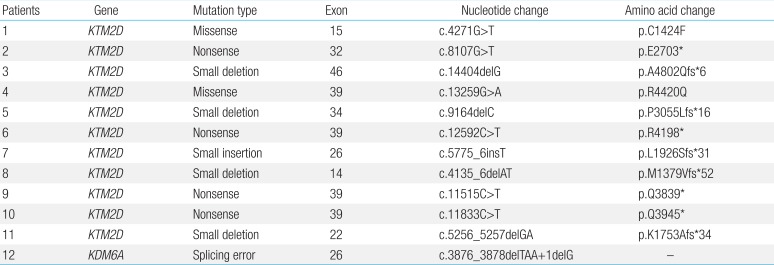
KS is first identified by a pediatrician after physical examination of the patient and review of his or her medical history. However, clinical diagnosis of KS by pediatricians is often challenging because the phenotype tends to evolve over time, and characteristic facial features, such as long palpebral fissures with everted lower lids, become more evident during childhood1). Currently, there are no established consensus diagnostic criteria for KS. Traditionally, five cardinal manifestations have been defined5): a 'peculiar face' (eversion of the lower lateral eyelid, arched eyebrows with the lateral one-third dispersed or sparse, a depressed nasal tip, and prominent ears; present in all patients), skeletal anomalies (deformed spinal column with or without sagittal cleft vertebrae and brachydactyly), dermatoglyphic abnormalities, mild to moderate mental retardation, and postnatal growth deficiency. Less frequent findings include visceral abnormalities, premature breast development in girls, and susceptibility to frequent infections. We recently phenotypically characterized 12 Korean patients with KS (Fig. 1)7). All patients showed the characteristic facial dysmorphism, including long palpebral fissures, eversion of the lateral portion of the lower eyelids, and a broad nasal root, and 92% displayed intellectual disabilities. Irrespective of racial differences, the suggested minimal criteria for diagnosis of KS are long palpebral fissures with eversion of the lateral portion of the lower eyelids, broad and arched eyebrows with lateral sparseness, short nasal columella with a depressed nasal tip, prominent or cupped ears, and developmental delay or mental retardation. Most patients with KS have several of these features that collectively produce an overall presentation suggestive of KS8).
Phenotypic characterization of Korean patients with Kabuki syndrome. The diverse phenotypes affecting multiple organ systems are noted.
What genes are associated with KS?
The genetic etiology of KS was not elucidated until 2010. At this time, Ng et al.2) reported that heterozygous mutations in KMT2D (MIM #602113) are the major genetic cause of KS. Subsequent mutation screenings in KS cohorts found a KMT2D nutation in 55.8% to 80% of patients69). Most mutations were private de novo mutations; familial occurrence with an autosomal dominant pattern has also been reported10). Most identified mutations are truncating, likely leading to haploinsufficiency of the KMT2D protein.
Heterozygous deletions in a second gene, KDM6A (MIM #300128), are another cause of KS10). In 2012, Lederer et al.11) reported the association of heterozygous whole or large partial deletion of KDM6A (observed in three patients) with a broad phenotypic spectrum, ranging from typical KS to a milder clinical presentation. A detection rate of 12.5% highlighted the usefulness of array-comparative genomic hybridization as a method for differential diagnosis in KMT2D-negative patients10). In 2013, point mutations in the KDM6A coding region in KMT2D mutation-negative KS individuals were also identified as KS-related12). In 2014, splice-site mutations in KDM6A were reported in Japanese and Korean patients713). Despite the rarity of KDM6A mutations in KS patients, KDM6A sequencing should be considered in all KMT2D mutation-negative KS patients. To date, the genetic basis of KS remains unknown in 20% to 45% of patients9), and further investigation is therefore needed. The role of histone modifications in KS also requires clarification, because the proteins encoded by both KMT2D and KDM6A are histone modifiers.
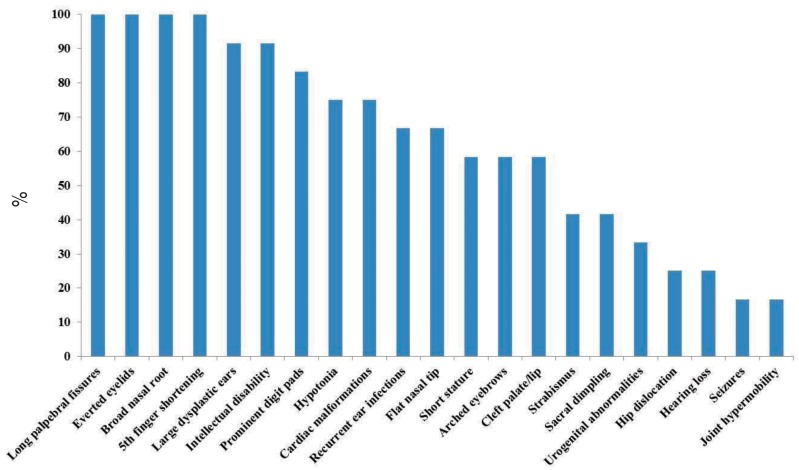
To date, 12 Korean patients with suspected KS in whom direct Sanger sequencing or exome sequencing was performed have been shown to express KMT2D (n=11) and KDM6A (n=1) variants (Table 1). All mutations were de novo, and each patient had a unique mutation. The 11 KMT2D mutations included five small insertions or deletions causing frameshifts, four nonsense mutations, and two missense mutations (Fig. 2). Ten (90.9%) were novel, and nine (81.8%) resulted in a truncated or shortened protein product. The mutations identified were relatively evenly distributed across exons 14 to 46 of the KMT2D gene, without a definite hot spot. However, four mutations were located in exon 39, which contains several regions that suggest the presence of a mutational hot spot (i.e., that encode long polyglutamine tracts)7).

Summary of the reported mutations in 12 Korean patients with Kabuki syndrome
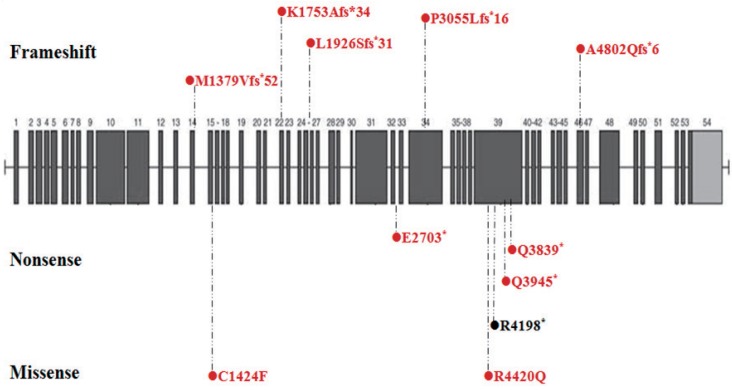
Distribution of KMT2D mutations in Korean patients with Kabuki syndrome. Schematic view of the KMT2D protein domains and all coding exons and the location of the KMT2D mutations. Dotted lines indicate the exonic location of the mutations identified in patients. Novel mutations are indicated by a red circle.
The high detection rate of KMT2D and KDM6A mutations (92.3 %) is expected owing to the strict criteria used to establish a clinical diagnosis6). Therefore, exhaustive mutation analysis of KMT2D and KDM6A may be a useful first step toward early diagnosis and counseling of Korean patients with KS. About 140 different mutations in the KMT2D gene have been reported in patients with KS (http://www.hgmd.org). Of these, 54 are nonsense mutations, 39 are small deletion mutations, 20 are missense mutations, 13 are small insertion mutations, nine are splicing mutations, three are small indel mutations, one is a gross deletion mutation, and one is a complex rearrangement. Including the 10 novel mutations reported in Korea, there are 57 nonsense, 42 small deletions, 22 missense, and 15 small insertions mutations (Fig. 3).
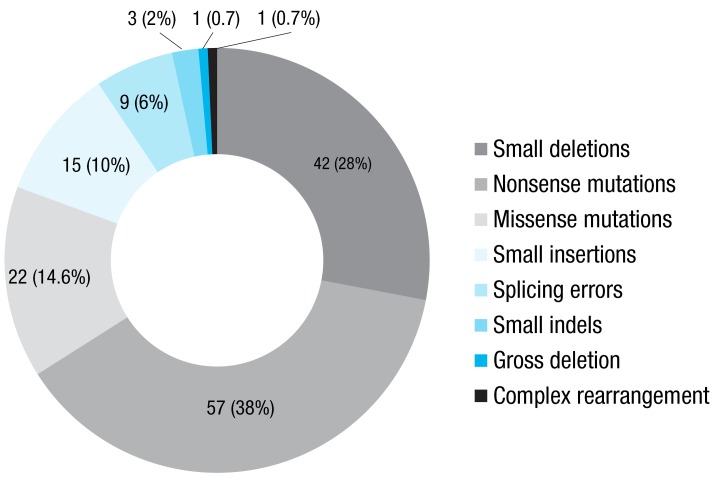
Frequencies of KMT2D mutations. The diagram shows the numbers and percentages of the KMT2D mutations listed in the Human Gene Mutation Database and 11 novel mutations identified in Korea.
Molecular mechanism of pathogenicity
KMT2D was identified and characterized in 1997. It encodes a histone H3 lysine 4 (H3K4)-specific methyltransferase needed for H3K4 di- and trimethylation, a hallmark of an active transcription state14). H3K4 methyltransferases act in stable multiprotein complexes that contain shared components as well as distinct components that denote the specific function of the complex10). KMT2D proteins contain a catalytic domain termed the SET domain, as do other histone methyltransferases, which are evolutionarily homologous to the Drosophila trithorax protein. The Su(var)3-9 and enhancer of zeste (SET) domains are also highly conserved in evolution15). The KMT2D complex ensures the transcription of constitutively expressed genes via H3K4 trimethylation. As part of the ASC-2 complex (ASCOM), KMT2D facilitates its binding to target genes16). Actions of ASCOM include the removal of repressive epigenetic marks, polycomb displacement, and positioning of activating methylation marks17).
KDM6A encodes a histone H3 lysine-27 (H3K27) demethylase14). It is important for general chromatin remodeling and interacts with KMT2D in a conserved SET-1-like complex that trimethylates H3K4. KDM6A and KMT2D interact with each other and are both trithorax-like proteins18). KMT2D methylates H3K4, and KMD6A demethylates H3K27, which activates transcription; conversely loss of KMT2D or KDM6A function represses transcription19). Little is known of the genes transcriptionally regulated by KMT2D complexes during development. Determination of the underlying pathogenesis of KS, including the functions of the proteins in these complexes during cell differentiation and growth is required10).
Clinical evaluation and management of patients with KS
Suggested evaluation and management procedures for patients with KS according to organ system are summarized in review articles59).
1. Body weight and growth
KS patients are typically born with normal growth parameters, but fail to thrive during infancy and show postnatal growth retardation because of feeding problems caused by poorly coordinated sucking and swallowing, as well as gastroesophageal reflux in some patients5). Feeding problems requiring a nasogastric tube or even gastrostomy have been documented in 65% to 74% of KS cases6). More than half the patients fail to thrive in infancy, and 57% have a body mass index in the overweight or obese range in middle childhood or adolescence20). Clinicians should inform the families of KS patients of the increased risk of obesity to promote awareness and early dietary intervention4).
2. Otolaryngological findings
The most common otolaryngological findings of KS include dysmorphic pinnae, otitis media, and hearing loss21). Middle ear infections occur in approximately 70% of patients and can lead to conductive hearing loss and hinder speech acquisition2). Conductive hearing loss appears to be the most frequent hearing impairment, followed by a sensorineural type of hearing loss. About 95% of KS patients have normal vestibular function10).
3. Ophthalmological findings
Ocular abnormalities have been described in 38% to 61% of KS patients10). The most common findings include ptosis, strabismus, and blue sclera1). Less common findings include coloboma of the iris and retina, refractive anomalies, Peter's anomaly, and congenital corneal staphyloma22). Severe visual impairment is rare in KS.
4. Cardiac malformations
Congenital heart defects are often observed in KS patients with frequencies of approximately 40% and 50% according to different literature reviews1). The most frequent malformations are atrial septal defects, ventricular septal defects, and aortic coarctation1). Coarctation of the aorta seems to be much more common in male patients than female patients8). Heart defects are equally frequent in patients with and without KMT2D mutations10). We recently reported hearing defects in 75.0% of KS patients; defects included Scimitar syndrome, coarctation of the aorta, ventricular septal defects, atrial septal defects, interrupted aortic arches, patent ductus arteriosus, and hypoplastic left heart syndrome7).
5. Gastrointestinal abnormalities
Gastrointestinal complications are very rare. Intestinal malrotation, abnormalities of the anus or rectum in the form of anal atresia, anovestibular fistula, and anteriorly placed anus have been reported1523). Previously described liver anomalies include biliary atresia, hepatic fibrosis, and sclerosing cholangitis23). Recently, a child with KS displayed inflammatory bowel disease24).
6. Urogenital abnormalities
Urinary tract anomalies are present in 30% to 40% of KS patients10) and include hydronephrosis, abnormal kidney position, renal hypoplasia or dysplasia, and fusion defects in the kidneys18). Renal malformations are significantly more common in patients with a KMT2D mutation10). Genital anomalies include cryptorchidism (25%), small penis (10%), and hypospadias1525). In our previous study, 36.3% of patients with a mutation in the KMT2D gene had renal duplication, multicystic dysplastic kidneys, ectopic kidneys, or a duplicated ureter7).
7. Endocrinological findings
Premature thelarche has been described in 41% of KS patients with a KMT2D mutation compared with 4% in mutation-negative patients9). It was not necessarily associated with other signs of precocious puberty10). Its underlying cause in KS patients is uncertain, but could be secondary to low hypothalamic sensitivity to the suppressive effects of sex hormones on gonadotropin secretion26). Rare findings in KS include hypothyroidism, hyperinsulinism hypoglycemia, diabetes insipidus, primary ovary dysfunction, and abnormal pituitary findings on magnetic resonance images320). Postnatal growth hormone deficiency was observed in 83% of KS patients5). Our recent study showed short stature in 50.0% of KS patients with KMT2D mutations7). Conidering the possibility of associated medical problems, such as skeletal and hematologic abnormalities, the benefits and side effects of growth hormone therapy for KS patients of short stature require further investigation in large patient cohorts10).
8. Skeletal findings
Skeletal abnormalities occur in about 80% of KS cases; they include rib anomalies, vertebrae malformations, scoliosis, cleft hand, and brachydactyly and/or clinodactyly of the fifth digit110). Cranial abnormalities include coronal and metopic synostosis, incomplete development of the frontal and/or maxillary sinuses, digital impression of the skull, and underdevelopment of the mastoid processes27).
9. Skin and connective tissue
Persistent fetal finger pads are a common and distinctive symptom of KS. Another common finding is an abnormal dermatoglyphic pattern, such as the absence of the digital triradius 'c' and 'd' and the presence of interdigital triradii, as well as hypothenar and interdigital ulnar loop patterns, which occur in almost all patients (96%) with KS7). Involvement of the connective tissue in KS has been proposed because of the combination of facial laxity, joint hyperlaxity, and joint dislocations10). Joint hypermobility is present in one-half to three-quarters of patients, and joint dislocations, particularly of the hips, patellae, and shoulders, are not uncommon1). Habitual joint dislocations in 32% of patients with KMT2D mutations compared with 9% in patients without KMT2D mutations have been reported28).
10. KS and cancer
To date, there are eight individuals with both KS and cancer worldwide, including two with neuroblastoma and one each with hepatoblastoma, low-grade fibromyxoid sarcoma, acute lymphocytic leukemia, Epstein-Barr virus (EBV)-positive Burkitt's lymphoma, EBV-negative Burkitt's lymphoma, and synovial sarcoma7829). Whether KS predisposes individuals to cancer remains uncertain10).
11. Immunologic/hematologic abnormalities
Patients with KS are vulnerable to infection in regions including the middle ear and upper airway tract. Decreased IgA and IgG levels were observed in 79% and 42% of patients, respectively30), and severe immunodeficiency with hypogammaglobulinemia requiring intravenous administration of intravenous immunoglobulin has been reported in several patients31). Scherer et al.32) described a 2-year-old girl with typical KS who developed acute lymphocytic leukemia. Autoimmune diseases, including idiopathic thrombocytopenic purpura and/or hemolytic anemia, and vitiligo are rarely seen in patients with KS33). Careful hematological investigation and surveillance is essential for early detection of immunologic or hematologic disease in patients with KS.
12. Neurological symptoms and development/behavior problems
Hypotonia and seizures may accompany the psychomotor development of KS patients10). Muscular hypotonia seems to be most striking in newborns and has been observed in 51% to 98% of patients310). Seizures mainly of the focal type originate predominantly in the temporo-occipital or frontocentral area and are well-controlled by antiepileptic drugs, resulting in a favorable prognosis34). We observed seizures in 16.7% of individuals with KMT2D mutations; all showed good responses to antiepileptic medications7). Patients with KS often exhibit nonspecific cerebral atrophy, ventriculomegaly, and microcephaly8). Several patients with autism or autistic-like behavior characterized by difficulties in both communication and peer interactions have been described135), suggesting that KS is a differential diagnosis in patients with facial dysmorphism presenting with autism spectrum disorder. Consistent with previous reports567), we observed varying degrees of intellectual disability in 91.7% of KS patients with KMT2D mutations36).
Genotype-phenotype correlation
Genetic changes causing KS have been recently discovered via exome sequencing, resulting in improved clinical diagnosis and more accurate genotype-phenotype correlations. However, correlations between mutation type and the phenotype of KS patients have yet to be definitively established. In a screen of 81 KS patients with or without KMT2D or KTM6A mutations, Miyake et al.28) observed the following. First, cleft lip/palate was more frequent in the mutation-positive group. Second, developmental delay and intellectual disabilities were detected in all patients with mutations but only in some patients without mutations. Third, blue sclera, lower lip pits, spine/rib abnormalities, hip joint dislocation, umbilical hernia, kidney abnormalities, cryptorchidism, liver/spleen abnormalities, premature thelarche, neonatal hyperbilirubinemia, and anemia were observed only in the mutation-positive group. Fourth, high arched eyebrows, short fifth fingers, and hypotonia in infancy were more common in individuals with KMT2D mutations compared with KDM6A mutations, whereas the opposite was true for short stature. Fifth, patients with truncating mutations more frequently had prominent ears and hypotonia and a more atypical facial appearance than patients without truncating-type mutations.
Patients with KDM6A mutations display a variety of phenotypes, ranging from typical KS to a milder clinical presentation1112). Banka et al.13) noted that patients with KDM6A mutations have hypotonia and feeding difficulties during infancy and poor postnatal growth and short stature. They suggested that hypertrichosis, long halluces, and large central incisors may be useful indicators of an underlying KDM6A mutation, even if facial dysmorphism is variable. Developmental delay and learning disabilities are generally moderate to severe in boys but mild to moderate in girls with KDM6A mutations13). We recently described a female patient with a KDM6A mutation who presented with typical facial dysmorphism, including long palpebral fissures, lateral eversion of the lower eyelid, short nasal septum, severe mental retardation, profound short stature, cardiac abnormality, recurrent infection, and Chiari malformation7).
Expert consultation and family genetic counseling
As KS is a multisystem disorder, people with KS may require various diagnostic and screening tests, assessments, referrals, and multidisciplinary interventions at different stages of their lives. Early diagnosis of the disease is crucial in regard to patient management of his or her associated medical problems and for prompt family and genetic counseling, and serves as a starting point for therapeutic interventions. Most reported KMT2D and KDM6A mutations are de novo mutations found in sporadic cases. In a few cases, an affected person is believed to have inherited the mutation from an affected parent. KS caused by mutations in the KMT2D gene is inherited in an autosomal dominant manner, whereas KS caused by mutations in the KDM6A gene is inherited in an X-linked inheritance manner. The KDM6A gene is located on the X chromosome, which is one of the two sex chromosomes. In women, a mutation in one of the two copies of the gene in each cell is sufficient to cause KS. In men, a mutation in the only copy of the gene causes the disorder. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Current research
Recently, a genetically engineered mouse model of KS with a heterozygous deletion in the gene encoding the lysine-specific methyltransferase 2D (Kmt2d) was developed. These mice (termed Kmt2d+/bGeo) have impaired methyltransferase function37). In vitro reporter alleles demonstrated reduced acetylation of histone H4 and histone H3 lysine K4 trimethylation (H3K4me3) in fibroblasts derived from Kmt2d+/bGeo embryos. Oral administration of the histone deacetylase inhibitor AR-42 to either young (1-2 months old) or adult (5-6 months old) Kmt2d+/bGeo mice eliminated the structural and functional deficits in the dentate gyrus in association with restoration of H3K4 trimethylation37). H3K4me3 deficiency in the dentate gyrus granule cell layer of Kmt2d+/bGeo mice correlated with reduced neurogenesis and hippocampal memory defects in an in vivo analysis, suggesting that KS could be treated by modulating the epigenetic machinery.
Conclusions
Considering the increasing number of patients newly diagnosed with KS owing to advanced comprehensive diagnostic services, clinicians need to be aware of KS and to look for a combination of abnormal medical conditions in patients with KS to prevent further complications. A greater understanding of patients with KS will allow physicians to better provide primary care to the patients and their families. It will also facilitate the detection of KS mutations, which depends on the accuracy of clinical diagnosis and the inclusion or exclusion of atypical cases. A population-based study is needed to accurately estimate the incidence of pediatric KS in Korea.
Acknowledgments
This research was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI12C0014).
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.