Nephrotic syndrome: what's new, what's hot?
Article information
Abstract
While the incidence of nephrotic syndrome (NS) is decreasing in Korea, the morbidity of difficult-to-treat NS is significant. Efforts to minimize treatment toxicity showed that prolonged treatment after an initial treatment for 2-3 months with glucocorticosteroids was not effective in reducing frequent relapses. For steroid-dependent NS, rituximab, a monoclonal antibody against the CD20 antigen on B cells, was proven to be as effective, and short-term daily low-dose steroids during upper respiratory infections reduced relapses. Steroid resistance or congenital NS are indications for genetic study and renal biopsy, since the list of genes involved in NS is lengthening.
Introduction
Nephrotic syndrome (NS) in children is a disease of glomerular filtration barrier failure, manifesting with severe proteinuria leading to hypoalbuminemia, hypercholesterolemia, and generalized edema. In this work, recent advances regarding idiopathic pediatric NS are reviewed.
Incidence
The prevalence of NS is 12-16 per 100,000 children1) with an annual incidence of 2-7 cases per 100,000 children. According to the Health Insurance Review and Assessment service (HIRA, http://www.hira.or.kr) of Korea, 4,342 patients younger than 20 years were treated for NS in 2009, while in 2013 the number of NS patients decreased to 2,795 (Fig. 1). Korean data show male to female ratio is 1.78-2.22 and prevalence of 37.0 in 2009 and 25.8 in 2013 per 100,000 children. Notably, prevalence according to HIRA may not be accurate because secondary NS, such as in Henoch-Schonlein nephritis, is not excluded, and the Korean Standard Classification of Disease (KCD-6), on which the HIRA data is based, is coded by individual users; nevertheless, prevalence of NS seems to be decreasing in Korea. Since it is known that NS is more common in developing countries, the decrease in NS in this country may be natural.
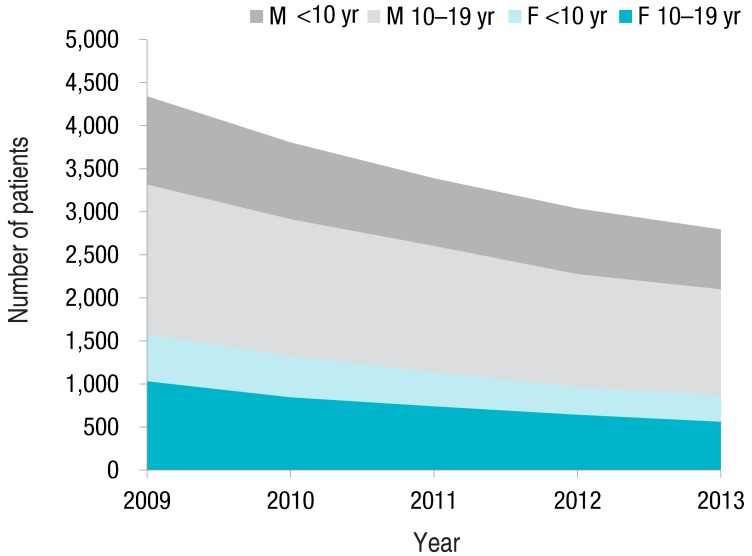
Number of patients younger than 20 years old treated for nephrotic syndrome from 2009 to 2013, according to the Health Insurance Review and Assessment service (http://www.hira.or.kr).
Clinical course
Twenty percent of idiopathic NS does not respond to steroid treatment. In addition to steroid resistance, frequent relapses and steroid dependence are concerns. Younger age, male gender, a history of atopy, longer time to first remission, a shorter time from remission to first relapse, and glucocorticoid receptor gene NR3C1 GR-9beta+TthIII-1 variants have been linked to frequent relapse and steroid dependence2345). One-third of patients do not have frequent relapses, and remission status at 6 months after initial presentation is a predictor of nonfrequent relapser6). In the 3 years after initial presentation, the relapse rate will decrease, and 80% will be in remission in 8 years6).
More than 15% of steroid-resistant NS (SRNS) will progress to end-stage renal disease (ESRD). Some of these will be found to have genetic NS (Table 1); detection of genetic causes in NS is an indication for cessation of immunosuppressive medications, because genetic NS does not respond to these, and inevitably progresses to ESRD. In contrast to idiopathic SRNS or focal segmental glomerulosclerosis (FSGS), with a recurrence rate of up to 50% after kidney transplantation (KT), genetic NS typically does not recur after KT7).

Causative genes of nephrotic syndrome/focal segmental glomerulosclerosis
Etiology
While the etiology of NS is not clear, conditions accompanying NS, such as leukemia/lymphoma and Kimura disease, suggest that the pathophysiology is immune-mediated.
1. Genetic causes of SRNS/congenital NS
Genetic causes need to be considered for patients with SRNS, congenital NS, family history of NS, and extrarenal involvement. Currently, there are 10 genes with names containing NPHS (autosomal recessive nephrotic syndrome), and 9 with FSGS (autosomal dominant FSGS, Table 1). Recent studies reported that 20%-30% of childhood-onset SRNS or congenital NS cases involved genetic causes89) (Fig. 2). While NPHS2 is the most common causative gene for European SRNS/congenital NS, in Northeastern Asia, the NPHS2 mutation is rare10); otherwise, genetic distribution is similar, with WT1 and NPHS1 (encoding nephrin) being the most common.

2. Circulating factor for FSGS
For SRNS, especially with recurrence after KT, a circulating factor has been hypothesized11). Soluble urokinase receptor (suPAR) has recently been suggested as this factor12). However, elevated suPAR did not induce proteinuria or FSGS in a recent study, and did not indicate FSGS clinically13) (Fig. 3).
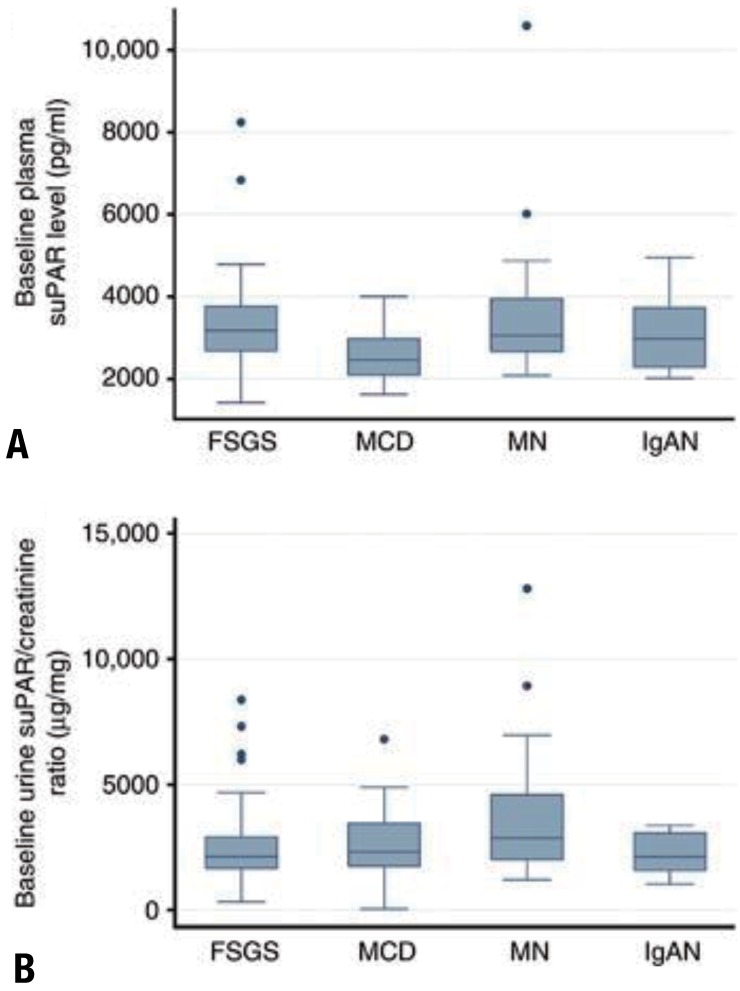
Baseline plasma and urine soluble urokinase-type plasminogen activator receptor (suPAR) concentrations by pathological diagnosis. (A) Baseline plasma suPAR concentration from 183 Nephrotic Syndrome Study Network (NEPTUNE) participants is plotted by diagnosis. Although minimal change disease (MCD) participants had a statistically significantly (P<0.01), lower median plasma suPAR concentration, as compared with all the other groups, this difference did not persist after adjustment for differences in estimated glomerular filtration rate. (B) Baseline urine suPAR/creatinine ratio from a 24-hour urine specimen in 173 NEPTUNE participants is plotted by diagnosis. Although membranous nephropathy (MN) participants had a statistically significantly greater median urine suPAR/creatinine ratio, as compared with focal and segmental glomerulosclerosis (FSGS) and immunoglobulin A nephropathy (IgAN) (P=0.01 and P=0.02, respectively), this difference did not persist after adjustment for differences in baseline urine protein. Adapted from Spinale JM, et al. Kidney Int 2015;87:564-74, with permission of International Society of Nephrology13).
3. Angiopoietin-like 4 as a mediator of persistent proteinuria and hyperlipidemia
Circulating angiopoietin-like 4 (Angptl4) level rises in response to proteinuria and increased serum free-fatty acids, via peroxisome proliferator-activated receptors (PPAR)α and PPARγ. It binds to αvβ5 integrin on glomerular endothelial cells, leading to a decrease in the severity of proteinuria14); in the circulation, Angptl4 inhibits lipoprotein lipase, leading to hyperlipidemia (Fig. 4). On the other hand, podocyte-producing hyposialylated Angptl4 is pro-proteinuric, suggesting the therapeutic potential of mutant Angptl4 as an antiproteinuric agent15).
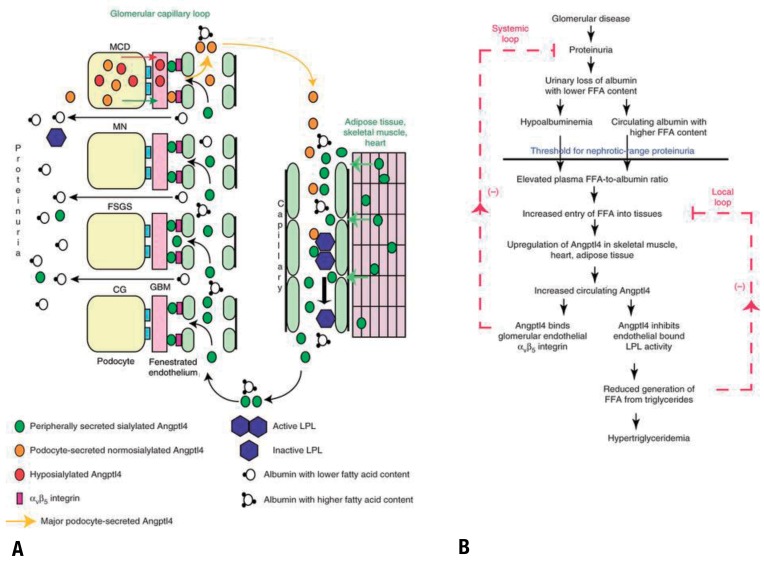
Pathobiology of circulating Angptl4 in nephrotic syndrome. (A) Diagram representing the production of circulating Angptl4 protein and its biological effects. The circulating, normosialylated form of Angptl4 is secreted from peripheral organs (mostly skeletal muscle, heart, and adipose tissue) in minimal change disease (MCD), membranous nephropathy (MN), focal and segmental glomerulosclerosis (FSGS), and collapsing glomerulopathy (CG). In addition, podocytes in MCD secrete a hyposialylated form of the protein that remains restricted to the kidney and induces proteinuria and a normosialylated form that enters the circulation. Circulating Angptl4 binds to glomerular endothelial αvβ5 integrin to reduce proteinuria, or inactivates endothelium-bound lipoprotein lipase (LPL) in skeletal muscle, heart, and adipose tissue, to reduce the hydrolysis of plasma triglycerides to free fatty acids (FFA), resulting in hypertriglyceridemia. Some Angptl4 and LPL are lost in the urine. (B) Schematic illustration of negative feedback loops in the link between proteinuria, hypoalbuminemia, and hypertriglyceridemia that are mediated by Angptl4 and FFA (unesterified fatty acids with a free carboxylate group). Plasma FFAs are noncovalently bound to albumin, and because of the preferential loss of albumin with low FFA content during proteinuria, albumin with higher FFA content is retained in circulation. As glomerular disease progresses and proteinuria increases, hypoalbuminemia develops, and the combination of high albumin-FFA content and lower plasma albumin levels increases the plasma ratio of FFAs to albumin. This increased available FFA enters the skeletal muscle, heart, and adipose tissue to induce up-regulation of Angptl4, mediated at least in part by Ppar proteins. Angptl4 secreted from these organs participates in two feedback loops. In the systemic loop, it binds to glomerular endothelial αvβ5 integrin and reduces proteinuria. In a local loop, it inhibits LPL activity in the same organs from which it is secreted, to reduce the uptake of FFAs, thereby curtailing the stimulus for its own up-regulation. Adapted from Clement LC, et al. Nat Med 2014;20:37-46, with permission of Macmillan Publishers Limited15).
4. Target antigen of membranous nephropathy
While membranous nephropathy (MN) accounts for only a small portion of pediatric NS, an interesting discovery is worth mentioning. In addition to neutral endopeptidase of antenatal MN16), an M-type phospholipase A2 receptor on the basal surface of podocytes has recently been identified as a target antigen in idiopathic MN (IMN), with autoantibodies in the circulation of more than 70% of patients with IMN17). For some patients, a thrombospondin type-1 domain-containing 7A was identified as a target antigen in MN18).
Diagnosis
1. Renal biopsy and diagnosis of FSGS
Pretreatment renal biopsy is necessary when the clinical profile of an NS patient is atypical. Therefore, if a patient is too young (<1 year) or too old (>12 years), has chronic infection, hypertension, azotemia, hematuria (≥30-49 red blood cells/high-power field)19), hypocomplementemia, or other findings implying another autoimmune disease, biopsy is indicated.
Diagnosis of FSGS on pathology may be missed due to sampling error; therefore, follow-up biopsy is required in some cases. On the other hand, diagnosis does not always imply primary FSGS, since FSGS may occur as a consequence of nephron mass reduction or renal damage. Secondary FSGS usually does not cause severe hypoalbuminemia, and electron microscopic examination often shows segmental foot process effacement, in contrast to the widespread foot process effacement in INS20) (Fig. 5). This differentiation is necessary because immunosuppressive treatment is not effective for secondary FSGS.
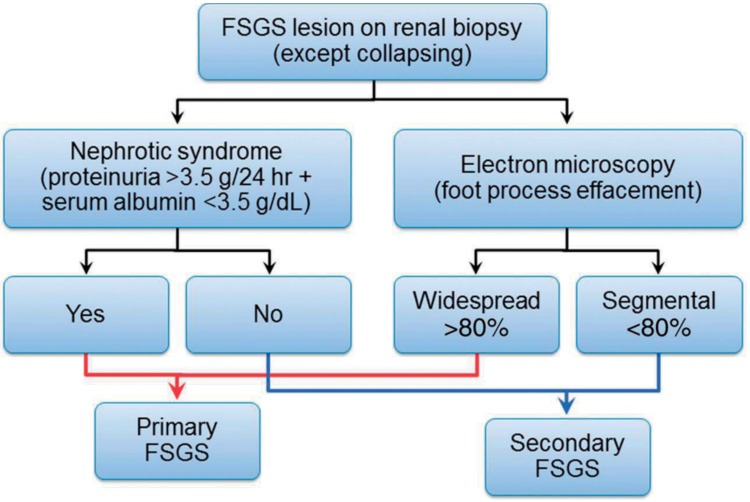
Proposed diagram to differentiate between primary and secondary focal segmental glomerulosclerosis (FSGS) based on clinical presentation and electron microscopic examination (collapsing FSGS is excluded). Adapted from Sethi S, et al. Nephrol Dial Transplant 2015;30:375-84, with permission of European Renal Association - European Dialysis and Transplant Assoc20).
2. Urinary marker of minimal change disease
Responsiveness to oral corticosteroid treatment is considered a clinical diagnosis of minimal change disease (MCD). Experimental models of NS have shown urinary CD80 levels are significantly elevated in MCD but not in others, and Ling et al. also reported similar findings in a clinical setting21).
Treatment
1. Duration of initial oral steroid treatment for steroid-sensitive NS
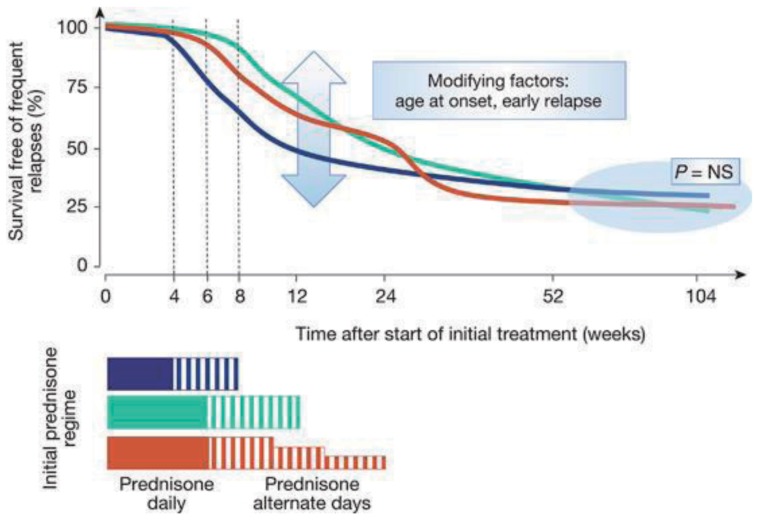
KDIGO (Kidney Disease: Improving Global Outcomes) published a guideline on glomerulonephritis in 2012 (http://kdigo.org/home/glomerulonephritis-gn/) including NS2223). It is recommended that oral corticosteroid therapy (prednisone, prednisolone, or deflazacort, 60 mg/m2/day or 2 mg/kg/day, up to 60 mg daily for 4-6 weeks; followed by 40 mg/m2 or 1.5 mg/kg every other day) be given for at least 12 weeks, and continued for 2-5 months, with slow tapering. However, recent, large, randomized prospective studies state otherwise32425) (Fig. 6); slow tapering did not reduce the relapse rate or incidence of frequent relapses242526). Therefore, overtreatment with long-term steroids is not recommended, until novel biomarkers indicating otherwise become available.
2. Steroid-sparing calcineurin inhibitors
For frequently relapsing or steroid-dependent NS (SDNS) and SRNS, a calcineurin inhibitor (CNI) is recommended as a steroid-sparing agent2223).
3. Rituximab
Rituximab (RTX) is a monoclonal antibody against the CD20 antigen on B cells, developed as a chemotherapeutic agent for B-cell lymphoma. The efficacy of this agent in NS was discovered when a patient with recurrent FSGS after KT, who had concurrent posttransplant lymphoproliferative disease, responded to RTX27). A few randomized, prospective trials on SDNS patients proved its efficacy, with a reduced relapse rate2829), and RTX is now considered a steroid- or CNI-sparing agent in SDNS30), enabling improved growth and reduction of obesity31). However, complications such as agranulocytosis (~10%) were reported32), and careful monitoring is therefore necessary after RTX treatment. RTX as an initial treatment of NS has not been reported.
While RTX treatment may maintain remission in the majority of SDNS patients, the effect often wanes over time, and many patients relapse 6 to 9 months after RTX treatment, with reappearance of CD19 (a marker of B cells)33). Maintenance with mycophenolate mofetil or an additional cycle of RTX treatment were successful in a few studies3034). Notably, anti-RTX antibodies may emerge with repetitive treatment; therefore, if there is a severe infusion reaction and CD19 persists despite RTX treatment, the presence of anti-RTX antibody should be considered35) (Fig. 7). In such cases, humanized anti-CD20 monoclonal antibody, such as ofatumumab can be considered, especially since ofatumumab was reported to be effective in RTX-resistant SRNS36).
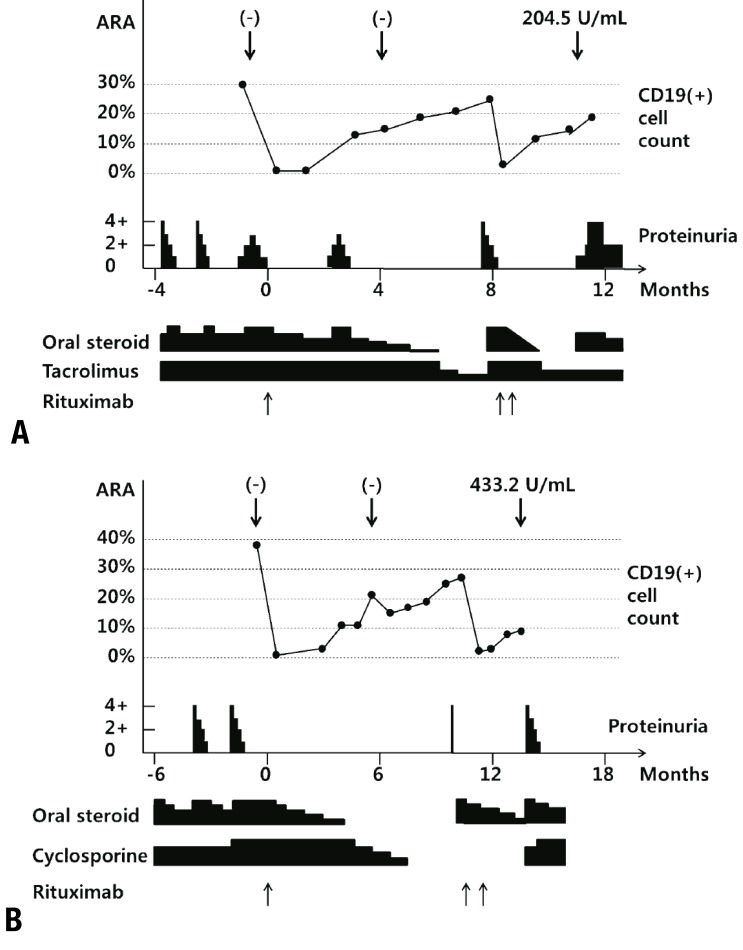
Clinical courses of patients who developed antirituximab antibodies. Proteinuria (lower bar, 0-4+), CD19-positive B-cell count (%, middle, line graph), and antirituximab antibody (ARA) titers at each point (down-pointing arrows), along the clinical course around rituximab treatments (up-pointing arrows below the graph). First treatment with rituximab was marked as month 0. Oral dosages of steroid and tacrolimus are represented as the height of the bar, below the graph. Adapted from Ahn YH, et al. Pediatr Nephrol 2014;29:1461-4, with permission of Springer International Publishing AG35).
For SRNS, RTX is effective in about 20% of patients37).
4. Galactose for SRNS
Galactose, a monosaccharide sugar, was shown to eliminate the proteinuric effect of FSGS-causing permeability factor (still of unknown identity) in an experimental model and some anecdotal cases38). In a prospective clinical trial on pediatric SRNS, galactose was not effective after 16 weeks39); however, reports from the Novel Therapies for Resistant Focal Segmental Glomerulosclerosis (FONT) trial (NCT00814255) state that some patients treated with oral galactose achieve a significant reduction in proteinuria during 24 weeks of therapy37).
5. Relapse prevention
Upper respiratory infection triggers relapse of NS, accounting for approximately 2/3 of relapses40). Three prospective studies showed a significant reduction in relapse rate with daily dosing of corticosteroids (conversion of alternate-day dosing to daily dosing or prednisolone of 0.5 mg/kg) for 5 to 7 days414243), during common viral infections. Supplementation with zinc (10 mg/day, the recommended dietary allowance) reduced relapses by 20%44).
6. Vaccination and supplements
NS patients are prone to infections caused by encapsulated bacteria, such as Streptococcus pneumoniae, Haemophilus influenzae, and group B streptococcus. Therefore vaccination is necessary; 23-valent pneumococcal polysaccharide vaccine is indicated in NS patients older than 2 years, with boosting every 5 years45). For those who had 13-valent pneumococcal vaccination, an interval greater than 8 weeks is required before 23-valent vaccination. Vitamin D or thyroid hormone replacement should be considered, as well as medication for dyslipidemia, if prolonged45).
Prognosis
1. Status in 10 years: frequently relapsing NS
In a long-term follow-up study of frequently relapsing nephrotic syndrome for more than 10 years, half of the patients had frequent relapses, and one-fifth were disease-free, with no relapse for 2 years. However, mortality or loss of kidney function was not identified49).
2. Steroid toxicity, quality of life
Adverse effects of long-term steroid treatment include short stature, osteoporosis, obesity, cataracts, hypertension, diabetes mellitus, and behavioral disturbances. Growth and bone mineral density were both negatively associated with the cumulative dose of glucocorticoid, and final height was significantly shorter in patients receiving >0.2 mg/kg/day doses of glucocorticoids50).
Long-term morbidity of pediatric NS, especially SDNS or SRNS, affects quality of life significantly, with impaired peer relationships, social functioning, and school performance, especially in those with longer disease duration51).
Conclusions
While NS is decreasing in Korea, the morbidity of difficult-to-treat NS is significant. Efforts to minimize toxicity showed that treatment with glucocorticosteroids prolonged beyond an initial 2-3 months was not effective in reducing frequent relapses. For SDNS, RTX, a monoclonal antibody against the CD20 antigen on B cells, was proven as effective, and short-term, daily low-dose steroid reduced relapses during upper respiratory infections. Steroid resistance or congenital NS are indications for genetic study and renal biopsy, since the number of genes involved in NS is increasing.
Acknowledgment
This work was supported by a grant (HI12C0014) from the Korea Healthcare Technology R&D Project, Ministry for Health and Welfare, Republic of Korea, and Basic Science Research Program, through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (NRF-2012R1A1A2006858).
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.
