Epilepsy in Korean patients with Angelman syndrome
Article information
Abstract
Purpose
The aim of this study was to investigate the natural history of epilepsy and response to anti-epileptic drug treatment in patients with Angelman syndrome (AS) in Korea.
Methods
We retrospectively reviewed the clinical records of 14 patients diagnosed with epilepsy out of a total of 17 patients with a genetic diagnosis of AS. These patients were seen at the Department of Pediatric Neurology at Severance Children's Hospital from March 2005 to March 2011.
Results
Fourteen (9 males and 5 females) subjects (82.3%) were diagnosed with epilepsy in AS. The most common seizure types were generalized tonic-clonic (n=9, 27%) and myoclonic (n=9, 27%), followed by atonic (n=8, 24%), atypical absence (n=4, 12%) and complex partial seizure (n=3, 9%). The most commonly prescribed antiepileptic drug (AED) was valproic acid (VPA, n=12, 86%), followed by lamotrigine (LTG, n=9, 64%), and topiramate (n=8, 57%). According to questionnaires that determined whether each AED was efficacious or not, VPA had the highest response rate and LTG was associated with the highest rate of seizure exacerbation. Complete control of seizures was achieved in 6 patients. Partial control was achieved in 7 patients, while one patient was not controlled.
Conclusion
Epilepsy is observed in the great majority of AS patients. It may have early onset and is often refractory to treatment. There are few reports about epilepsy in AS in Korea. This study will be helpful in understanding epilepsy in AS in Korea.
Introduction
Angelman syndrome (AS) is a neurogenetic disorder characterized by severe mental retraction, speech disorder, stereotyped jerky movement and an overall happy disposition accompanied with frequent laughter1-3). The incidence of AS is estimated to be between 1 in 10,0004). The most common genetic subtype causing AS is a maternal deletion of chromosome 15q11-13 (68 to 75%), followed by unknown (10 to 20%), UBE3A mutations (8 to 11%), uniparental disomy (UPD, 2 to 7%), and imprinting defects (ID, 2 to 5%)5).
About 90% of these patients have epileptic seizures6,7). The age of seizure onset is often between 1 and 3 years. Many seizure types, both generalized and focal, have been reported, including atypical absence, myoclonic, atonic, and generalized tonic-clonic (GTC) seizures8,9). Epilepsy associated with AS can be self-limited, particularly after puberty10-12). However, many patients experience more intractable seizures as well as repeated episodes of status epilepticus (SE).
There are only a few reports about epilepsy in patients with AS in Korea13). In this article, we analyzed 14 patients with epilepsy in AS and characterized their clinical features, particularly those pertaining to epilepsy.
Materials and methods
We retrospectively reviewed the clinical records of 14 patients (82.3%) with epilepsy out of 17 patients that had been given the genetic diagnosis of AS, who visited Severance Children's Hospital from March 2005 to March 2011. The genetic study was carried out using an analysis of the methylation pattern of the 15q11-13 region of chromosome 15 and fluorescence in situ hybridization (FISH) in order to determine the type of underlying mutation.
Demographic data including age and gender were collected from medical records while clinical data including epilepsy were collected from questionnaires as well as from medical records. We studied the characteristics of the epilepsy, age of onset, frequency and type of seizures, and the type of treatment patients received. We also analyzed the characteristics of the electroencephalography (EEG) results initially and during follow-up.
Results
Of the 17 patients (9 males and 8 females) with the genetic diagnosis of AS, 3 patients (18%) were determined by maternal deletion of chromosome 15q11-13 by FISH, but 14 patients (82%) were determined by abnormal methylation of maternal allele only, and further evaluation to identify the underlying mechanism (deletion, UPD, ID or UBE3A gene mutation) was not done. Of the 14 patients with epilepsy in AS reviewed in this study, 9 were males and 5 were females. The mean age of the patients at the time of the study was 9.7 years (range, 2.7 to 24 years), and mean duration of the follow-up was 6.7 years (range, 2.1 to 23 years). Their clinical characteristics are shown in Table 1.
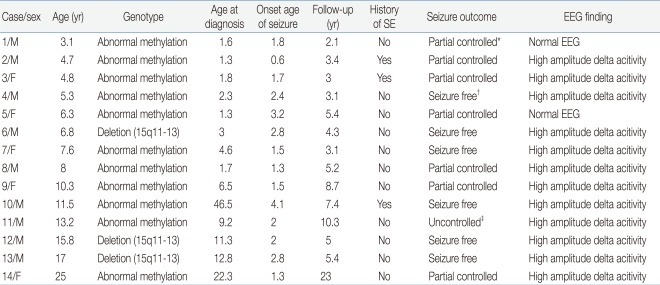
Clinical Characteristics in Patients with Angelman Syndrome
The mean age at diagnosis of AS was 6.5 years, with an age range between 1.4 years and 22.4 years. Twelve patients were diagnosed with epilepsy under 3 years of age (86%). In 2 patients over the age of three years presenting with epilepsy, the mean age of diagnosis of epilepsy was 3.6 years.
Age of seizure onset was a mean of 1.9 years (range, 7 months to 4.0 years). The most frequent types of initial seizure were GTC (n=4, 29%) and atonic (n=4, 29%), followed by myoclonic (n=3, 21%), complex partial seizure (n=2, 14%) and atypical absence (n=1, 7%) (Fig. 1). During the follow-up period, the most common seizure types were GTC (n=9, 27%) and myoclonic (n=9, 27%), followed by atonic (n=8, 24%), atypical absence (n=4, 12%) and complex partial seizure (n=3, 9%). Multiple seizure types were observed in 10 patients (71%). Three patients presented episodes of SE.
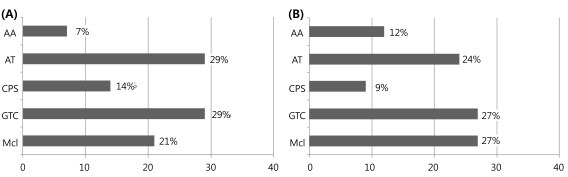
The types of initial seizure and during follow-up period. (A) Initial seizure type. (B) Sezure type during follow-up. AA, atypical absence seizure; AT, atonic seizure; CPS, complex partial seizure; GTC, generalized tonic-clonic seizure; Mcl, myoclonic seizure.
Ten patients were able to walk alone, whereas 4 patients were not. The mean age when the patients started walking was 3.8 years (range, 2 to 6.5 years). At the time of the study, patients had an average of 2.4 current antiepileptic drug (AED) medications, with 14% and 86% having tried monotherapy and multiple medications. The most commonly prescribed AED was valproic acid (VPA, n=12, 86%) followed by lamotrigine (LTG, n=9, 64%), topiramate (TPM, n=8, 57%), levetiracetam (LEV, n=5, 36%), zonisamide (ZNS, n=5, 36%), clonazepam (CLZ, n=2, 14%) and phenobarbital (Pb, n=2, 14%) (Table 2).
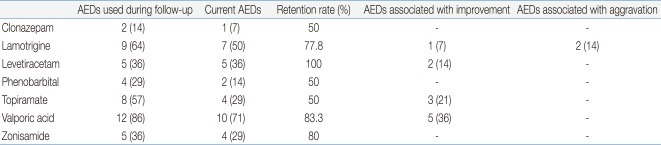
History of Antiepileptic Drugs (AEDs) Treatment in Angelman Syndrome
In our series, the EEG patterns initially showed a typical AS pattern characterized by slow waves of high voltage in 71% (n=10) of patients. Normal EEG patterns were observed in 14% (n=2) of patients. Family members of the study subjects were asked whether each AED was efficacious or not in controlling the episodes of epilepsy. VPA (n=5, 36%) had the highest response rate of being efficacious, followed by TPM (n=3, 21%), LEV (n=2, 14%), and LTG (n=1, 7%). LTG (n=2, 14%) was associated with the highest rate of seizure exacerbation.
No patients tried non-pharmacologic therapies such as dietary therapy, vagal nerve stimulation or surgical intervention for their epilepsies. Complete control (seizure free more than a year) of seizures was achieved in 6 patients. Partial control (seizure more than once a year, but under once a month) was achieved in 7 patients, while 1 patient was not controlled (seizure more than once a month). Control of seizures was achieved at a mean age of 7.5 years (range, 3.6 to 14.1 years).
Discussion
In more than 90% of patients with a clinical diagnosis of AS, genetic testing can demonstrate a molecular mechanism that causes lack of expression of the UBE3A gene. To date, 4 genetic mechanisms have been described for this syndrome. The known genetic mechanisms are deletion (del[15q11-13])14-16), paternal UPD17), ID18), and UBE3A mutations19,20). Maternal deletion (15q11-13) occurs in 75 to 80% of patients. In our study, three patients (17.6%) were determined by maternal deletion of chromosome 15q11-13 by FISH and 14 patients (82.4%) were determined by abnormal methylation of the maternal allele. Most commercially available DNA methylation analysis tests cannot distinguish between AS resulting from a deletion, UPD, ID and UBE3A mutation21). Further testing is required to identify the underlying molecular mechanism such as FISH or array CGH to determine the deletion of chromosome 15q11-13. If the FISH or array CGH analysis is normal, analysis of DNA polymorphisms on chromosome 15 can distinguish between UPD and an ID. In our study, except 3 patients diagnosed as maternal deletion (15q11-13), further evaluation for the determination of more precise genetic subtypes was not performed.
Epileptic seizures occur in 80% of patients that have AS22). Similarly in our study, 82.3% of patients with AS were diagnosed as having epilepsy. Onset of seizures is often before 3 years of age, mostly between 1 and 3 years10,11,23,24). In our study, mean age of seizure onset was 1.9 years, and 12 patients (86%) under 3 years of age were diagnosed with epilepsy. As reported by Matsumoto et al.12), the atypical absence and myoclonic seizures were the most frequent seizures at onset with previous studies demonstrating myoclonic seizure as a predominant seizure type25). In our experience, most common types of initial seizure were GTC (n=4, 29%) and atonic (n=4, 29%), followed by myoclonic (n=3, 21%), complex partial seizure (n=2, 14%) and atypical absence seizure (n=1, 7%). These seizures (myoclonic and atypical absence seizures) may have been under reported in this study because myoclonic seizures are easily misunderstood as behavioral problems in AS, particularly at early stages. Also, atypical absence seizures are easily misunderstood as complex partial seizures because this information has been collected by questionnaires based on a retrospective method. A prospective study with a larger group of patients and correlation of video EEG monitoring are required for more accurate conclusions.
The percentage of those with multiple seizure types (71%) was somewhat higher what has been described in previous reports26,27). Many different types of seizures have been reported, both generalized and focal. The most common seizure types previously reported were atonic, GTC, atypical absence and myoclonic seizure28-30). In our study, most common types of seizure during follow-up were GTC and myoclonic, followed by atonic, atypical absence seizure. GTC seizures showed a tendency to occur with fever in particular. In previous reports, it was also observed that febrile seizures often precede the diagnosis of AS31) and that even moderate temperature increases show a triggering effect24). Excluding febrile convulsion, myoclonic, followed by atonic and atypical absence seizures were the most common seizure types. Laan et al.29) reported SE in 36.1% of their patients, and Sugimoto et al.32) reported SE in 75% of patients. In our study, SE occurred in 21% (n=3), and the main seizure type in SE was myoclonic seizure.
Ten patients were able to walk alone (group A), whereas 4 patients were not. The mean age at which the patients started walking was 3.8 years (range, 2 to 6.5 years). Although four patients were older than the mean age of walking, two patients (group B, 4.6 years and 4.4 years) had a higher probability to walk alone in comparison with the other two patients (group C, 12.9 years and 10 years) because they were younger than the age to walk alone of the latest patient (6.5 years). Group C showed the worst seizure control as uncontrol and partial control in each patient. Group B showed better seizure control than group C as partial controls in all 2 patients. Group A showed best seizure control as complete control in 6 patients, and partial control in 4 patients. There are reports that the deletion phenotype is generally linked to a more severe clinical picture in that 95% of patients manifest more severe seizure patterns and mental retardation33). However, there is no report about the direct correlation between seizure control and development in AS. Further study including their genetic subtype, state of development and degree of seizure control with a larger group of patients is required.
Epilepsy in AS appears to improve around puberty32), or during adulthood11). Similarly, in our study, 3 patients after puberty showed better seizure control (complete control in 2 patients and partial control in 1 patient) than patients before or in the period of puberty (complete control in 4 patients, partial control in 6 patients and uncontrolled in 1 patient).
There are specific EEG patterns in AS patients which appear in isolation or in different combinations. They are similar in patients both with and without seizures. In childhood, the three characteristic patterns are 1) persistent rhythmic 4 to 6 Hz activity, 2) prolonged runs of rhythmic triphasic 2 to 3 Hz activity, maximal over the frontal regions and normally mixed with spikes or sharp waves, and 3) spikes mixed with 3 to 4 Hz component, mainly posteriorly and facilitated by eye closure34). In our study, the EEG initially showed the typical AS pattern characterized by runs of high-amplitude slow waves that were more prominent in the frontal region mixed with spike and slow waves multifocal activity in 12 patients (86%) and normal pattern in 2 patients. The interictal EEG patterns were not related to the type of initial seizures and EEG abnormalities persisted even when seizures were controlled.
Earlier reports suggested a decreasing frequency of epileptic seizures with age11,29). In our study, control of seizures was achieved at a mean age of 7.5 years (range, 3.5 to 14 years). However, the high prevalence (77%) of subjects with epilepsy that was medically refractory appears consistent with nearly all previously published reports. In our study, the 57% of patients that had medically refractory epilepsy was somewhat lower than in previous reports.
Many AEDs are used to treat seizures in individuals with AS, but there is no agreement on optimal seizure medication, although VPA, LTG, TPM, LEV, and CLZ are more commonly used34). Carbamazepine (CBZ), oxcarbamazepine, and vigabatrine have been noted to cause paradoxical aggravation of seizures in some individuals with AS9), but their use in AS is not absolutely contraindicated. As reported by Thibert et al.26), three newer AEDs that were most commonly prescribed (TPM, LTG, and LEV) had better perceived efficacy and tolerability than CBZ and Pb. Experience with the newer AEDs is very limited. In our study, the drugs most frequently used included VPA (n=12, 86%) with a retention rate of 83.3% in the patients, LTG (n=9, 64%) with retention rate of 77.8%, and TPM (n=8, 57%) with retention rate of 50%. At present, 2 patients receive a single drug (LEV, TPM), and 12 patients receive multiple drug therapy.
Some children with uncontrollable seizures have been placed on a ketogenic diet, and occasionally vagal nerve stimulation has been used. As reported by Thibert et al.26), the classic ketogenic diet appeared to be most efficacious. In our study, no patients tried nonpharmacologic therapies such as dietary therapy, vagal nerve stimulation or surgical intervention for their epilepsies. According to questionnaires on whether AEDs were efficacious or not, VPA (n=5, 36%) had the highest response rate of being efficacious, followed by TPM (n=3, 21%), LEV (n=2, 14%) and LTG (n=1, 7%). LTG (n=2, 14%) was associated with the highest rate of seizure exacerbation.
The limitation of this study was that a more precise genetic subtype of each patient was not evaluated. Lossie et al.21) reported patients with UPD are less severely affected so that the diagnosis is often suspected later. Minassian et al.33) reported patients with imprinting anomalies had an even milder course with fewer seizures and better communication skills. Further study of precise genotypes was required to evaluate correlations between clinical phenotypes and genotypes.
In conclusion, epilepsy is very common in AS and typically refractory to medication. Our study will be helpful in understanding epilepsy in AS patients in Korea. Further characterization of epilepsy in AS along with advances in genetic analyses will hopefully lead to a better understanding of the pathogenesis of epilepsy in AS. Further studies with a larger population on effective treatment of epilepsy in AS and improvements in the management of problems such as behavior, communication, learning, motor impairment, and sleep disturbances in AS are also needed.
Acknowledgment
This work was supported by National Research Foundation grant funded by the Korea government (MEST) (2010-0020353).