A case of de novo duplication of 15q24-q26.3
Article information
Abstract
Distal duplication, or trisomy 15q, is an extremely rare chromosomal disorder characterized by prenatal and postnatal overgrowth, mental retardation, and craniofacial malformations. Additional abnormalities typically include an unusually short neck, malformations of the fingers and toes, scoliosis and skeletal malformations, genital abnormalities, particularly in affected males, and, in some cases, cardiac defects. The range and severity of symptoms and physical findings may vary from case to case, depending upon the length and location of the duplicated portion of chromosome 15q. Most reported cases of duplication of the long arm of chromosome 15 frequently have more than one segmental imbalance resulting from unbalanced translocations involving chromosome 15 and deletions in another chromosome, as well as other structural chromosomal abnormalities. We report a female newborn with a de novo duplication, 15q24-q26.3, showing intrauterine overgrowth, a narrow asymmetric face with down-slanting palpebral fissures, a large, prominent nose, and micrognathia, arachnodactyly, camptodactyly, congenital heart disease, hydronephrosis, and hydroureter. Chromosomal analysis showed a 46,XX,inv(9)(p12q13),dup(15)(q24q26.3). Array comparative genomic hybridization analysis revealed a gain of 42 clones on 15q24-q26.3. This case represents the only reported patient with a de novo 15q24-q26.3 duplication that did not result from an unbalanced translocation and did not have a concomitant monosomic component in Korea.
Introduction
Distal trisomy, or duplication of 15q, is an extremely rare chromosomal disorder in which the end portion of the long arm of the 15th chromosome (15q) appears three times (trisomy), rather than twice, in cells of the body. Since Fujimoto et al.1) reported on duplication of distal 15q in 1974, at least 50 cases of duplication of regions 15q21-15q26.3 have been identified. The range and severity of symptoms and physical findings may vary from case to case, depending upon the length and location of the duplicated portion of chromosome 15q. However, there are consistent and recognizable clinical phenotypes with partial duplication for distal 15q, which are characterized by minor craniofacial anomalies, congenital heart disease, mental retardation, and genital anomalies, particularly in affected males2). Most reported cases have resulted from unbalanced translocations or deletions involving chromosome 15 and another chromosome3). We report on a girl with a de novo duplication, 15q24-qter, with breakpoints that differ from those that have been described previously and did not result from unbalanced translocation or deletion. Bacterial artificial chromosome (BAC) clone-based-array comparative genomic hybridization (aCGH) analysis revealed a gain of 42 clones on 15q24-qter. Thus, the patient was diagnosed as having de novo duplication of chromosome 15; 15q24-q26.3. This case represents the only reported patient in Korea with a 15q24-qter duplication or trisomy that did not result from an unbalanced translocation and did not have a concomitant deletion.
Case report
A female infant was born at 37+3 weeks of gestation following a repeat caesarean section performed in a local obstetrics clinic. She was the second child of healthy, phenotypically normal, and non consanguineous Korean parents. Her mother was 34 years old, and her father was 33 years old. There were no reported drug or teratogenic exposures during pregnancy and normal fetal activity was reported. The patient's Apgar scores were 5 and 8 at 1 and 5 minutes. She weighed 3,940 g (>90 percentile), was 52 cm long (>90 percentile), and had a head circumference of 36 cm (>90 percentile) at birth. She had cyanosis and irregular respiration, as well as peculiar facial morphology, and multiple anomalies, including craniofacial limbs, were detected after birth. Thus, she was transferred to the Neonatal Intensive Care Unit of Pusan Paik Hospital, Inje University College of Medicine.
She had a large anterior fontanel with wide sutures, a narrow, asymmetric face, with down-slanting palpebral fissures, and a large, prominent nose, pointed chin, and micrognathia, low set dysmorphic ears with poorly formed helices (Fig. 1); she also had long, tapering fingers, and slender hands, with a drumstick appearance of the terminal phalanges, and narrow feet, with broad big toes (Fig. 2). A grade II systolic murmur on the left sternal border and coarse breath sounds were heard. Chest radiograph showed atelectasis and ground glass opacities in both lung fields. Patent ductus arteriosus and atrial septal defects were detected on echocardiogram. Abdominal ultrasound examination showed bilateral hydronephrosis and hydroureter.

Photographs of the Face. (A) Narrow asymmetric face with down-slanting palpebral fissures, and large prominent nose, pointed chin and micrognathia are shown in the photograph of the face. (B) Low set dysmorphic ears with poorly formed helices, micrognathia and retrognathia are shown in lateral view of patient's face.

Photographs of hand and foot. (A) Arachnocamptodactyly; long tapering fingers, and slender hand with drumstick appearance of the terminal phalanges are shown. (B) Narrow foot with broad big toe.
1. Cytogenetic analysis
Standard chromosomal analysis of peripheral blood by Giemsatrypsin banding showed additional chromosomal material on the long arm of one chromosome 15 and in all cells examined. Results from propositus demonstrated 46,XX,inv(9)(p12q13),dup(15) (q24q26.3). The karyotype of the father was normal, 46, XY, while that of the mother showed 46,XX,inv(9)(p12q13) (Fig. 3).
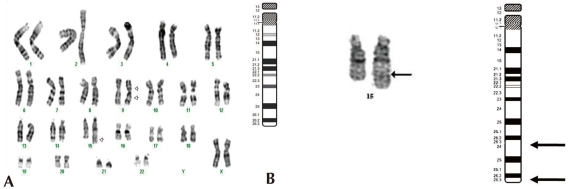
GTG banded Karyotypes of the lymphocytes from the patient. (A) The Result of karyotyping was 46,XX,inv(9) (p12q13),dup(15)(q24q26.3). The arrows indicate the abnormal chromosomes. Duplication of the distal chromosome 15q with inversion of chromosome 9 were detected. (B) Chromosome 15 ideogram and partial karyotype of the patient indicating the extent of duplicated region (q24->q26.3) (arrow). The abnormal chromosome is on the right.
2. aCGH analysis
We have developed a microarray for comparative genomic hybridization utilizing genomic clones from chromosome 15q for characterization of genomic rearrangements. The array accurately localized all breakpoints associated with gains or losses on 15q. We used the MAC Array Karyo 4000 (Macrogen Inc., Seoul, Korea) spotted with 4,030 human (BAC) clones, which covered the entire human genome at an average interval of 0.83 Mb. These BAC clones were selected from genome databases archived at the gene ontology website (http://www.geneontology.org). Array-based CGH was performed as reported. Images (16-bit) of fluorescence intensity for spots were captured from each array slide with a Gene Pix 4000A scanner and the ratio value was calculated using MAC viewer software (Macrogen Inc.). Average log2 Cy3/Cy5 signal ratios of triplicate BAC clones were calculated for each sample, and ±0.25 log2 ratio was used as a threshold for defining copy number increases (gains) and decreases (losses). BAC clone-based-aCGH analysis revealed a gain of 42 clones on 15q24-q26.3. Thus, the patient was diagnosed as having de novo duplication 15 q24-q26.3 (Fig. 4).
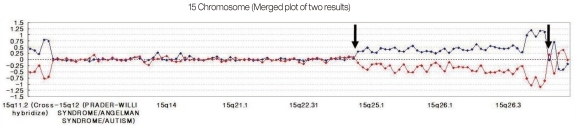
The ratio plots from array comparative genomic hybridization. Normalized data in which the test sample was labeled with cyanine 3 are shown in blue, while that in which the test sample was labeled with cyanine 5 are shown in red. A gain of a particular clone is manifested as a positive (upward) deviation of the blue signal from modal value 0.25 and a negative (downward) deviation of the red signal from -0.25 for same clone. Conversely, a loss of a clone shows the opposite pattern. A forty two - clone gain on 15q24-q26.3 (between arrows) are found.
Discussion
Partial trisomy, or duplication of the long arm of chromosome 15, represents a very rare and heterogenous group of chromosomal aberrations, which were first described by Fujimoto et al.1) in 1974. Common phenotypic findings in the distal duplication 15q syndrome are consistent and are characterized by down slanting palpebral fissures, a large, prominent nose, facial asymmetry, ptosis, micrognathia, congenital heart disease, severe mental deficiency, and genital anomalies, including cryptorchidism and hypogonadism (in affected male), and hand anomalies (arachnodactyly and camptodactyly)2,4). In the majority of patients, duplication of 15q is concomitant with translocation or deletion. Breakpoints in distal 15q trisomy occur most often between bands 15q21 and 15q235,6). Van Allen et al.7) reported on families with breakpoints at 15q15 and 15q26, respectively. In contrast to the above cases, our patient has a duplication of the mid-distal part of the long arm of chromosome 15. Zollino et al.8) reviewed the most detailed reports on a total of 32 patients with the 15q duplication and divided them into two groups, based on their trisomic extent: one group (26 patients) had trisomy for 15q21-qter, and the other group (6 patients) had trisomy for 15q25qter. They compared and described the clinical phenotypes between duplication 15q21-24 and duplication 15q25-26qter. Both groups shared mental retardation, a prominent nose, a long, narrow, and asymmetric face, severe scoliosis with chest deformity, a pointed chin, and micrognatia, hypotonia, congenital heart defects, and renal abnormalities. Clinical features typical for trisomy for 15q25-qter included tall stature (in 4 of 6 patients), macrocephaly, craniosynostosis, and broad thumbs/big toes. Clinical features specific for duplication of 15q21-24 included microcephaly and normal prenatal growth2). The present report provides further evidence for a specific phenotype related to duplication of 15q25 or 26-qter with macrosomia at birth; overgrowth, macrocephaly, and broad thumbs and big toes, but craniosynostosis. Over growth is rarely associated with chromosomal anomalies. Overgrowth has been reported as well in association with duplication of distal 15q. The cause of overgrowth and tall stature has been thought to be the triplication of the insulin-like growth factor 1 receptor (IGF1R) gene located on 15q26.3. Insulin-like growth factors (IGF) and their receptors have been implicated in pre- and postnatal growth. IGF1R is a transmembrane heterotetramer, located on 15q25-q26.3, which mediates the mitogenic effect of IGF1 and IGF29). Faivre et al.10) suggested that overgrowth might be causally related to a dosage excess of the IGF1R gene. Of particular interest, overgrowth has also been reported in patients with tetrasomy of chromosome 15q25-qter, as well as in some patients with larger trisomy 15q22qter. Also, a gene for macrocephaly is likely to be located in this region. Of particular interest, a large family with autosomal recessive syndrome of macrocephaly, multiple epiphyseal dysplasia, and distinctive facial features has been recently reported, and a gene locus was identified on 15q26.2411).
In contrast, patients with 15q-monosomy, including a deletion of distal 15q and a ring chromosome 15, showed some distinct clinical findings. Most of these patients had intrauterine growth retardation (IUGR), microcephaly, abnormal face and ears, micrognathia, a highly arched palate, renal abnormalities, lung hypoplasia, failure to thrive, and developmental/mental retardation12). Roback et al.13) reported on an infant with del(15)(q26.1qter) and a girl with r(15) (p12q26.3), respectively; both patients had IUGR that was associated with loss of an allele of the (IGF1R) gene.
Most cases of partial trisomy, or duplication of a segment of distal 15q, were the result of unbalanced translocations. The second chromosome involved in the reciprocal translocation has varied; chromosomes 2, 7, 8, 11, 13, 15 (Robertsonian translocation), and 21 have been reported6). Other cases of 15q duplication have been the result of direct or inverted duplication14,15). However, in this case, there were no concomitant translocations or deletions. This case represents the only reported patient with a de novo 15q24-q26.3 duplication that did not result from an unbalanced translocation and did not have a concomitant monosomic component in Korea. In our case, BAC clone-based-aCGH analysis revealed a gain of 42 clones on 15q24-qter. Thus, the patient was diagnosed as having de novo 46,XX,inv(9)(p12q13),dup(15)(q24q26.3) chromosome complement. Microarray-based CGH using large-insert clones is useful for detection of deletions or duplications that are greater than about 10 kb, but below the level of detection by karyotype analysis. Microarray CGH enables assessment of hundreds or even thousands of sites in a single hybridization, thus providing a whole-genome scan for rearrangements. Array CGH has been used for detection of constitutional chromosomal abnormalities. Recent studies utilizing chromosome 15-specific BAC microarrays of varying resolution have provided more detailed molecular characterization of chromosome 15 rearrangements16).
The majority of idic(15q) chromosomes are comprised of symmetrical arms with four copies of the breakpoint 1 to breakpoint 5 region. Patients with less common breakpoints that are not distinguished by routine cytogenetic methods have been more accurately characterized by array analysis. This microarray provides a detailed characterization for chromosomal abnormalities involving 15q and is useful for more precise genotype-phenotype correlations17).
We have developed a microarray for comparative genomic hybridization utilizing genomic clones from chromosome 15q for characterization of abnormal regions. The array accurately localized all breakpoints associated with gains or losses on 15q. The results confirmed the location of breakpoints associated with interstitial duplications. Partial duplication of distal 15q due to de novo pure duplication is very rare18). This case represents the only reported patient in Korea with a 15q24-qter duplication or trisomy that did not result from an unbalanced translocation and did not have a concomitant monosomic component. Cytogenetic analysis of the parents demonstrated that no other chromosomal aberrations, except for inv(9)(p12q13), were present in the mother, who was phenotypically normal. Inversion of chromosome 9; inv(9)(p12q13) was demonstrated in both the patient and the mother. Pericentric inversion of chromosme 9 is one of the most common structurally balanced chromosomal variations and has been found in normal populations, and does not appear to be pathogenic with regard to congenital anomalies19). The present case necessitates the continuing delineation of phenotype-karyotype correlations and phenotypic consequences of chromosome 15q abnormalities.