A case of McKusick-Kaufman syndrome
Article information
Abstract
McKusick-Kaufman syndrome (MKS) is an autosomal recessive multiple malformation syndrome characterized by hydrometrocolpos (HMC) and postaxial polydactyly (PAP). We report a case of a female child with MKS who was transferred to the neonatal intensive care unit of Seoul National University Children's Hospital on her 15th day of life for further evaluation and management of an abdominal cystic mass. She underwent abdominal sonography, magnetic resonance imaging, genitography and cystoscopy which confirmed HMC with a transverse vaginal septum. X-rays of the hand and foot showed bony fusion of the left third and fourth metacarpal bones, right fourth dysplastic metacarpal bone and phalanx, right PAP and hypoplastic left foot with left fourth and fifth dysplastic metatarsal bones. In addition, she had soft palate cleft, mild hydronephroses of both kidneys, hypoplastic right kidney with ectopic location and mild rotation, uterine didelphys with transverse vaginal septum and low-type imperforated anus. She was temporarily treated with ultrasound-guided transurethral aspiration of the HMC. Our patient with HMC and PAP was diagnosed with MKS because she has two typical abnormality of MKS and she has no definite complications of retinal disease, learning disability, obesity and renal failure that develop in Bardet-Biedl syndrome, but not in MKS until 33 months of age. Here, we describe a case of a Korean patient with MKS.
Introduction
The phenotype of McKusick-Kaufman syndrome (MKS) was initially described by McKusick in the Amish population1). The phenotypic triads of MKS comprise hydrometrocolpos (HMC), postaxial polydactyly (PAP) and congenital heart disease (CHD)2). For females without a family history in the non-Amish population, HMC with distal vaginal agenesis or a transverse vaginal membrane and PAP are considered sufficient clinical evidence of MKS, and the diagnosis is usually made at birth3). Here, we describe a case of MKS which has rarely been reported in Korea.
Case report
This 4.3 kg, female newborn infant was born by cesarean section at 37 weeks of gestation to a 40-year-old Gravida 2 Para 2 Korean mother at a local obstetric clinic. Apgar scores were 8 and 9 at 1 and 5 minutes. Her prenatal history and laboratory studies were normal. Prenatal sonograms showed no abnormalities at 35 weeks of gestation. After birth, the baby had abdominal distension and multiple gross anomalies. Abdominal sonography showed mild hydronephroses of both kidneys and 9.5 cm cystic mass at the suprapubic area. She was transferred to a university hospital near her home. She underwent abdominal magnetic resonance imaging (MRI) which showed HMC and a transverse vaginal septum. She was temporarily treated with percutaneous drainage (which was removed due to dysfunction after 2 days) and ultrasound-guided transurethral aspiration of the HMC. The drained amount from the abdominal cyst was 60 to 100 mL. She was transferred to the neonatal intensive care unit of Seoul National University Children's Hospital on her 15th day of life for further evaluation and management. Her height was 51 cm (50 to 75 percentile), weight 3,680 g (50 to 75 percentile), head circumference 36 cm (50 to 75 percentile), and abdominal circumference 35.5 cm. Physical examination revealed a hairy forehead and ear, bilateral preauricular pits, highly arched palate and soft palate cleft. Grade 1 to 2 systolic murmurs were audible on the left anterior precordium. Distended abdomen, right PAP and syndactyly (fourth and fifth), left foot deformity, and anteriorly placed stenotic anus were also present (Fig. 1). She also had asymptomatic pyuria and bacteriuria. However, she had neither bacteremia nor sepsis. The neonatal screening test and the tandem mass test were normal. She did not undergo molecular genetic analysis.
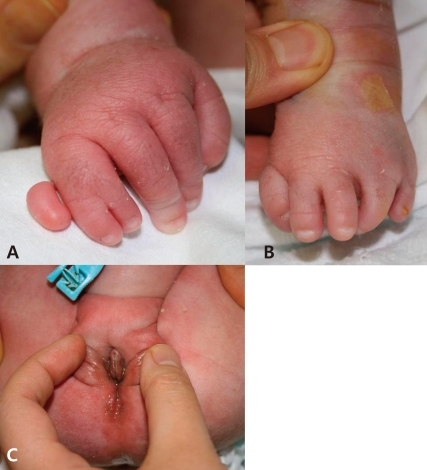
A) Postaxial polydactyly and syndactyly are seen in the right hand. B) A left foot deformity is seen. C) An anteriorly placed stenotic anus is seen.
X-rays of the hand and foot showed bony fusion of the left third and fourth metacarpal bones, right fourth dysplastic metacarpal bone and phalanx, right PAP and hypoplastic left foot with left fourth and fifth dysplastic metatarsal bones (Fig. 2). Abdominal sonography revealed HMC, uterine didelphys with a transverse vaginal septum, bilateral hydronephroureteroses, hypoplastic right kidney (2.4 cm) with ectopic location and mild rotation, and nonvisualization of both ovaries. Anterior displacement of the collapsed bladder was also noted. On non-enhanced abdominal MRI (outside film), HMC was seen as a large cystic mass with septation, transverse vaginal septum and severe hydronephroureteroses, and both ovaries were not visible (Fig. 3). She underwent genitography and cystoscopy which confirmed HMC with a transverse vaginal septum. Brain sonograms revealed no significant abnormalities. Echocardiography showed a patent foramen ovale with left to right shunt and peripheral pulmonary stenosis on the right pulmonary artery which had a velocity of 1.9 meters per second. On ophthalmologic evaluation, the cornea, lens and retina were normal.
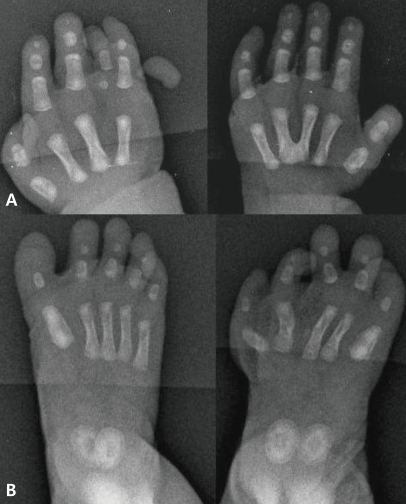
A) Right-hand postaxial polydactyly, fourth dysplastic metacarpal bone/phalanx, and bony fusion of the left third and fourth metacarpal bones are seen. B) A hypoplastic left foot with the fourth and fifth dysplastic metatarsal bones is seen.
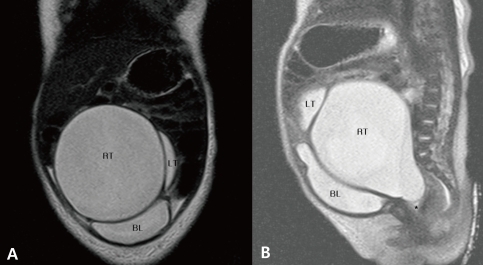
A) The distal part of hydrometrocolpos is divided into right and left cysts. B) A transverse vaginal septum (linear lesion above ' * ' marking) is seen. RT, right; LT, left; BL, bladder. *, vaginal cavity.
She was temporarily treated with ultrasound-guided transurethral aspiration of the HMC. The size of the cyst waxed and waned. However, urination was good and there was no renal dysfunction. A pediatric urologist planned to follow up the patient with sonography, urinalysis, blood urea nitrogen (BUN) and creatinine for checking hydronephroureteroses at 2 months of age. For the low-type imperforated anus, a pediatric surgeon planned a jump-back operation at 3 months of age. A pediatric plastic surgeon planned to follow up the patient for soft palate cleft and operation of the right PAP at 1 year of age. She was discharged from the hospital and revisited the outpatient clinic 16 days after discharge. Follow-up abdominal sonography showed decrease of the size of the HMC. But the patient could not undergo operation for the low-type imperforated anus (anteriorly placed stenotic anus), PAP and soft palate cleft, because of economic problems. Recently, she was scheduled again for operation to solve these problems.
Discussion
The first case of MKS in Korea was reported by Kong et al.4) in 1992. MKS has been unrecognized or underreported in Korea because only 1 case has been reported so far. The first case of MKS was a full-term infant and at 14 days of age. She had HMC, PAP of both feet, hydronephroureteroses and umbilical hernia. She had a distal transverse vaginal septum. She underwent vaginal plastic surgery. Her final diagnosis was MKS with HMC and PAP. They did not describe ophthalmologic findings and differential diagnosis. We should consider a differential diagnosis of MKS and Bardet-Biedl syndrome (BBS) because HMC and PAP are commonly present in both disease entities3). BBS is a genetically heterogeneous autosomal recessive condition characterized by retinitis pigmentosa, PAP, central obesity, learning disability, hypogonadism and renal anomalies5-8). Although hypogenitalism, PAP and renal anomalies may be apparent at birth in BBS, the other clinical features develop in childhood and the average age of diagnosis is 9 years8). The complications of BBS are severe, and almost all patients become blind by the age of 20 years7). A newborn with HMC and PAP can be diagnosed with either MKS or BBS because manifestations specific to BBS are age dependent9). However, it is important to discriminate between the 2 syndromes based on the complications of retinal disease, learning disability, obesity and renal failure that develop in BBS, but not in MKS3).
Our patient had no clinical characteristics of BBS except for a hypoplastic right kidney (2.4 cm) with ectopic location and mild rotation. Level of BUN and creatinine were normal. Although she did not undergo regular follow-up, she had no problems with urination. On admission, ophthalmologic evaluation was normal. She is healthy and at 29 months of age now. Her pronunciation was incorrect probably because of her soft palate cleft, but she could sing along to songs. Her family did not notice that she had learning disabilities. Her weight is under the normal value. It is possible that she is underweight because she has severe constipation that required finger enema and abdominal compression every 2 or 3 days and she does not eat enough food.
Some authors reported patients with BBS who had structural abnormalities of the female genitourinary tract without HMC in the neonatal period3,7,10,11). Complete agenesis of the vagina has been described in females with BBS12) but not in MKS patients in whom only agenesis of the distal third of the vagina or a transverse vaginal membrane was present3). It is possible that abnormalities of the uterus, ovaries and fallopian tubes or complete absence of the vagina are important clinical features which permit discrimination between MKS and BBS phenotypes in early life3). In our case, the patient had a uterine didelphys with a transverse vaginal septum. It is thought that the double-blind cysts were formed by HMC of the uterine didelphys. For a correct diagnosis, follow-up sonography was needed. Abdominal sonography and nonenhanced abdominal MRI (outside film) revealed no visualization of either ovary. However, it was difficult to find the ovaries in the abdominal cavity because these tests were performed in the neonatal period and because there was a huge cystic mass beside the ovaries. It was also difficult to find the ovaries by nonenhanced MRI. Follow-up sonography or MRI was needed to precisely evaluate the ovaries. In non-Amish MKS patients, 2-3, 3-4 and 4-5 syndactyly are frequently found, whereas 2-3 syndactyly is the most common in BBS patients8). Our patient had 4-5 syndactyly.
MKS is caused by mutations in the MKKS gene on chromosome 20p13). Both MKS and BBS are inherited in an autosomal recessive trait, and both syndromes are caused by mutations in the MKKS gene14). However, mutations in the MKKS gene are found in only 4 to 11% of the unselected BBS patients14). To date, 14 BBS genes, BBS1 to BBS14, have been identified, accounting for over 75% of mutations in BBS families (BBS1 to BBS14, namely BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, TTC8, BBS9, BBS10, TRIM32, BBS12, MKS1, CEP290)15). Molecular genetic analysis may be useful for treating and counseling females with HMC and PAP3), but there was no formal gene examination of the MKS and BBS in Korea.
Slavotinek and Biesecker3) reviewed the reported cases of MKS and BBS presenting with HMC and PAP early in life. They recommended that the current clinical care should include monitoring for the complications of BBS.
Our patient with HMC and PAP was diagnosed with MKS because she has two typical abnormality of MKS and she has no definite evidence of complications of retinal disease, learning disability, obesity and renal failure that develop in BBS, but not in MKS3) until 33 months of age. We need to monitor the complications of BBS in the future. Here, we describe a case of a Korean patient with MKS.